NGS Data Analysis: Data Formats and Workflow Managers
 Bhagesh Hunakunti
Bhagesh Hunakunti
Introduction
Next-generation sequencing (NGS) has revolutionized the field of genomics by allowing researchers to generate vast amounts of sequencing data quickly and cost-effective manner. However, the analysis of NGS data can be complex and challenging, especially for those who are new to the field. One of the most significant challenges in NGS data analysis is understanding the various data formats and workflow managers used in the process.
NGS data is typically generated in various formats, each with its advantage and limitations. These data formats include FASTQ, BAM, VCF, and BED, among others. Understanding these formats and their uses is crucial in ensuring accurate and efficient data analysis.
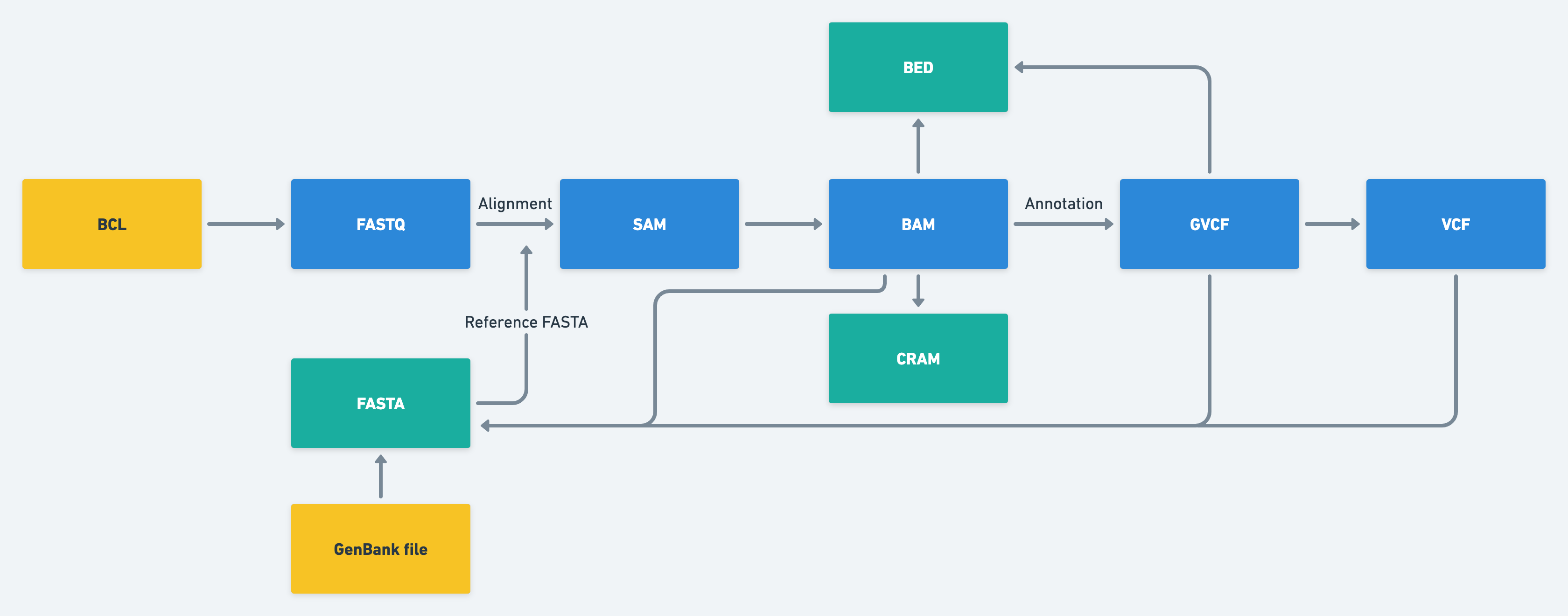
In addition to data formats, NGS data analysis also requires the use of workflow managers to streamline the analysis process. Workflow managers, such as Snakemake, Nextflow, and Cromwell, help automate the analysis process, making it more efficient and reproducible.
In this article, we will explore the different data formats used in NGS data analysis and the various workflow managers used to automate the process. This article will serve as a guide to help researchers navigate the complex landscape of NGS data analysis, from data processing to downstream analysis.
NGS Data Formats
Well, I guess bioinformaticians just wanted to keep things interesting! They figured, why settle for one boring data format when you can have a whole bunch to choose from?
It's like a data format buffet - you get to sample a little bit of everything. Just make sure you don't get indigestion from all those files!
There are multiple reasons why a variety of data formats are required in NGS analysis:
Efficiency: Different data formats are optimized for different types of data and analyses, which allows for more efficient processing and storage of large amounts of data.
For example, the BAM format is designed for storing aligned reads and associated metadata, while the VCF format is designed for storing genomic variations. Each format is tailored to a specific purpose, allowing for more efficient data processing and analysis.
Compatibility: Different software tools and analysis pipelines may require different data formats as input or output, which can lead to the use of multiple data formats in a single analysis workflow. This is particularly true in the rapidly evolving field of NGS analysis, where new tools and methods are constantly being developed and tested.
Standardization: Data formats such as FASTQ and BAM have become standard formats for storing raw sequencing data and aligned reads, respectively, and are widely used by the NGS community. Standardization helps to ensure consistency and interoperability between different software tools and analysis pipelines.
Data sharing: Different data formats may be required for sharing data with collaborators or submitting data to public repositories. For example, the GenBank format is commonly used for sharing annotated DNA and protein sequences, while the VCF format is commonly used for sharing genomic variation data.
In summary, the use of multiple data formats in NGS analysis is driven by the need for efficiency, compatibility, standardization, and data sharing. While it may seem daunting at first, familiarity with common data formats and their use in different analysis pipelines can greatly facilitate NGS data analysis.
FASTA/Multi-FASTA
The FASTA format is widely used in the fields of bioinformatics and biochemistry as a text-based format to represent either nucleotide or amino acid (protein) sequences. It employs single-letter codes to represent the nucleotides or amino acids. The format allows for the inclusion of sequence names and comments preceding the sequences. Originally from the FASTA software package, it has now become a near-universal standard in the field of bioinformatics.The simplicity of the FASTA format makes it easy to manipulate and parse sequences using text-processing tools and scripting languages.
A sequence in FASTA format begins with a greater-than-character > followed by a single-line description of the sequence. The next lines immediately following the description line provide the sequence representation, with each amino acid or nucleic acid represented by a single letter. Typically, these lines are no more than 80 characters in length.
>BA000007.3 Escherichia coli O157:H7 str. Sakai DNA, complete genome
AGCTTTTCATTCTGACTGCAACGGGCAATATGTCTCTGTGTGGATTAAAAAAAGAGTCTCTGACAGCAGC
TTCTGAACTGGTTACCTGCCGTGAGTAAATTAAAATTTTATTGACTTAGGTCACTAAATACTTTAACCAA
TATAGGCATAGCGCACAGACAGATAAAAATTACAGAGTACACAACATCCATGAAACGCATTAGCAC...
A multi-fasta file is a text file containing multiple sequences in FASTA format. Each sequence begins with a header line, which starts with the greater-than symbol >, followed by a brief description of the sequence, and then the sequence itself. The sequences are separated by one or more blank lines.
Multifasta files are commonly used in bioinformatics to store and process large numbers of sequences. They can be created by concatenating individual FASTA files or by exporting sequences from a database or software tool in the FASTA format. They can be processed using a wide range of software tools, including sequence alignment, motif detection, and phylogenetic analysis programs.
The NCBI has established a standard for the unique identifier used in the header line for sequences, called the SeqID. This standard enables a sequence obtained from a database to be labelled with a reference to its database record. NCBI tools such as makeblastdb and table2asn recognize the database identifier format.
The following list outlines the NCBI FASTA-defined format for sequence identifiers:
| Type | Format(s) |
| local (i.e. no database reference) | lcl |
| GenInfo backbone seqid | bbs |
| GenInfo backbone moltype | bbm |
| GenInfo import ID | gim |
| GenBank | gb |
| EMBL | emb |
| PIR | pir |
| SWISS-PROT | sp |
| patent | pat |
| pre-grant patent | pgp |
| RefSeq | ref |
| general database reference (a reference to a database that's not in this list) | gnl |
| GenInfo integrated database | gi |
| DDBJ | dbj |
| PRF | prf |
| PDB | pdb |
| third-party GenBank | tpg |
| third-party EMBL | tpe |
| third-party DDBJ | tpd |
| TrEMBL | tr |
The FASTA input does not allow for blank lines within the sequence representation. The sequences should follow the standard IUB/IUPAC amino acid and nucleic acid codes, with the exception that lower-case letters can be used and will be converted to upper-case. Additionally, a single hyphen or dash can represent a gap of indeterminate length, while U and * are acceptable letters in amino acid sequences (see below).
Before submitting a request, any numerical digits present in the query sequence must be removed or replaced by appropriate letter codes such as N for an unknown nucleic acid residue or X for an unknown amino acid residue.
The supported nucleic acid codes include:
| Nucleic Acid Code | Meaning | Mnemonic |
| A | Adenine | A |
| C | Cytosine | C |
| G | Guanine | G |
| T | Thymine | T |
| U | Uracil | U |
| (i) | inosine (non-standard) | - |
| R | A or G (I) | puRine |
| Y | C, T or U | pYrimidines |
| K | G, T or U | Ketones |
| M | A or C | aMino groups |
| S | C or G | Strong interaction |
| W | A, T or U | Weak interaction |
| B | not A (i.e. C, G, T or U) | B comes after A |
| D | not C (i.e. A, G, T or U) | D comes after C |
| H | not G (i.e., A, C, T or U) | H comes after G |
| V | neither T nor U (i.e. A, C or G) | V comes after U |
| N | A C G T U | Nucleic acid |
| - | a gap of indeterminate length | - |
The supported amino acid codes consist of 22 standard amino acids and three special codes:
| Amino Acid Code | Meaning |
| A | Alanine |
| B | Aspartic acid (D) or Asparagine (N) |
| C | Cysteine |
| D | Aspartic acid |
| E | Glutamic acid |
| F | Phenylalanine |
| G | Glycine |
| H | Histidine |
| I | Isoleucine |
| J | Leucine (L) or Isoleucine (I) |
| K | Lysine |
| L | Leucine |
| M | Methionine/Start codon |
| N | Asparagine |
| O | Pyrrolysine (rare) |
| P | Proline |
| Q | Glutamine |
| R | Arginine |
| S | Serine |
| T | Threonine |
| U | Selenocysteine (rare) |
| V | Valine |
| W | Tryptophan |
| Y | Tyrosine |
| Z | Glutamic acid (E) or Glutamine (Q) |
| X | any |
| * | translation stop |
| - | a gap of indeterminate length |
The table below displays the common filename extensions used for text files containing FASTA formatted sequences along with their meanings:
| Extension | Meaning | Notes |
| fasta, fa | Generic FASTA | Any generic fasta file. See below for other common FASTA file extensions. |
| fna | FASTA nucleic acid | Used generically to specify nucleic acids. |
| ffn | FASTA nucleotide of gene regions | Contains coding regions for a genome. |
| faa | FASTA amino acid | Contains amino acid sequences. A multiple protein fasta file can have the more specific extension mpfa. |
| frn | FASTA non-coding RNA | Contains non-coding RNA regions for a genome, in DNA alphabet e.g. tRNA, rRNA |
BCL
The BCL (Base Call) file format is a binary file format used to store raw data from Illumina sequencing instruments.
A BCL file typically contains four types of data:
Base call data - this data represents the actual base calls that were made during sequencing. Each base call is represented by a pair of bits, with '00' representing an 'A' base call, '01' representing a 'C' base call, '10' representing a 'G' base call, and '11' representing a 'T' base call.
Quality score data - this data represents the quality of each base call, with higher scores indicating a greater confidence in the accuracy of the base call.
Signal data - this data represents the raw fluorescence signal intensities captured during sequencing.
Index data - this data represents the index sequence used to demultiplex the sequencing data, allowing for identification of which sample each read belongs to.
BCL files are typically generated in pairs, one file containing data from the forward read and another file containing data from the reverse read. These files are usually compressed using gzip or bzip2 compression to reduce file size, and the file extension for BCL files is typically '.bcl'.
The BCL file format is used in the early stages of processing raw sequencing data and contains information about the intensity of fluorescent signals emitted by nucleotides during sequencing. After the BCL files are generated, they are typically converted into other file formats, such as FASTQ, for downstream analysis.
BCL files are an important intermediate format in Illumina sequencing data analysis, but they are not typically directly analyzed by researchers. Instead, BCL files are typically converted into other more accessible file formats for downstream analysis.
FASTQ
The FASTQ file format is a text-based file format used in bioinformatics to store both sequencing reads and their associated quality scores. It is a widely used format for storing sequencing data from a variety of platforms, including Illumina, Ion Torrent, and PacBio.
A FASTQ file contains a series of records, each representing a single read. Each record consists of four lines:
Sequence identifier line - this line starts with a
@symbol followed by a unique identifier for the read.Sequence line - this line contains the actual nucleotide sequence of the read.
Quality score identifier line - this line starts with a
+symbol and may optionally contain the same unique identifier as the sequence identifier line.Quality score line - this line contains a string of characters representing the quality scores for each base call in the corresponding sequence line.
The quality score line contains ASCII-encoded characters, with each character representing the quality score for the corresponding base call in the sequence line. The quality scores are typically represented as Phred scores, which are logarithmically transformed probability values. A higher Phred score indicates a higher confidence in the accuracy of the corresponding base call.
Here's an example of a FASTQ record:
@HWI-ST1276:182:C0AN3ACXX:2:1101:14768:1948
GAGCGGTCTCGCGATCGTCAGCGCGCGGTTGCGCGCGCGCGCGCGCGCG
+
B@BFFFFHHHHHJJJJJJJJJJIJIJJJJIJJIJJJJJJIJIJIIJJJIH
In this example, the sequence identifier line starts with @HWI-ST1276:182:C0AN3ACXX:2:1101:14768:1948, which is a unique identifier for this read.
The second line contains the nucleotide sequence, GAGCGGTCTCGCGATCGTCAGCGCGCGGTTGCGCGCGCGCGCGCGCGCG.
The third line starts with a + symbol and may optionally contain the same unique identifier as the sequence identifier line.
The fourth line contains the quality scores for each base call in the corresponding sequence line, which are represented as ASCII-encoded characters. The quality of a base call in a FASTQ file is represented by a byte, which ranges from 0x21 (lowest quality, represented by the character '!' in ASCII) to 0x7e (highest quality, represented by the character '~' in ASCII). The quality value characters are ordered in increasing quality, as shown below:
| ASCII Character | Score | |
| ! | 0 | |
| " | 1 | |
| # | 2 | |
| $ | 3 | |
| % | 4 | |
| & | 5 | |
| ' | 6 | |
| ( | 7 | |
| ) | 8 | |
| * | 9 | |
| + | 10 | |
| , | 11 | |
| - | 12 | |
| . | 13 | |
| / | 14 | |
| 0-9 | 0-9 | |
| : | 36 | |
| ; | 37 | |
| < | 38 | |
| \= | 39 | |
| \> | 40 | |
| ? | 41 | |
| @-Z | 0-23 | |
| [ | 42 | |
| \ | 43 | |
| ] | 44 | |
| ^ | 45 | |
| _ | 46 | |
| ` | 47 | |
| a-z | 0-25 | |
| { | 48 | |
| 49 | ||
| } | 50 | |
| ~ | 51 |
Note that the ASCII characters with scores 0-62 are inclusive of the printable ASCII character set.
In the original Sanger FASTQ file format, long sequences and quality strings were split over multiple lines, similar to how it's done in FASTA files. However, this made parsing more complicated due to the use of "@" and "+" characters as markers, which could also appear in the quality string. Multi-line FASTQ files and parsers are less common now, as most sequencing is carried out using short-read Illumina sequencing with typical sequence lengths of around 100bp.
The Illumina software generates a systematic identifier for sequencing reads that provides useful information about the origin of the read. In older versions of the pipeline (prior to 1.4), the identifier takes the form of:
@HWUSI-EAS100R:6:73:941:1973#0/1
In this format, the identifier consists of the unique instrument name, the flowcell lane, the tile number within the flowcell lane, the x- and y-coordinates of the cluster within the tile, an index number for a multiplexed sample (if applicable), and the member of a pair (if the reads are paired-end or mate-pair reads).
HWUSI-EAS100R | the unique instrument name |
|---|---|
6 | flowcell lane |
73 | tile number within the flowcell lane |
941 | 'x'-coordinate of the cluster within the tile |
1973 | 'y'-coordinate of the cluster within the tile |
#0 | index number for a multiplexed sample (0 for no indexing) |
/1 | the member of a pair, /1 or /2 (paired-end or mate-pair reads only) |
Newer versions of the pipeline (1.4 and above) use a slightly different format:
@EAS139:136:FC706VJ:2:2104:15343:197393 1:Y:18:ATCACG
In this format, the identifier includes the unique instrument name, the run ID, the flowcell ID, the flowcell lane, the tile number within the flowcell lane, the x- and y-coordinates of the cluster within the tile, the member of a pair (if applicable), whether the read is filtered (did not pass), an even number if any of the control bits are on, and the index sequence (if applicable).
EAS139 | the unique instrument name |
|---|---|
136 | the run id |
FC706VJ | the flowcell id |
2 | flowcell lane |
2104 | tile number within the flowcell lane |
15343 | 'x'-coordinate of the cluster within the tile |
197393 | 'y'-coordinate of the cluster within the tile |
1 | the member of a pair, 1 or 2 (paired-end or mate-pair reads only) |
Y | Y if the read is filtered (did not pass), N otherwise |
18 | 0 when none of the control bits are on, otherwise it is an even number |
ATCACG | index sequence |
It is worth noting that in some cases, the index sequence may be replaced with a sample number if an index sequence is not explicitly specified for a sample in the sample sheet. For example, the following header might appear in a FASTQ file belonging to the first sample of a batch of samples:
@EAS139:136:FC706VJ:2:2104:15343:197393 1:N:18:1
Overall, the FASTQ file format is an important format for storing sequencing data, as it contains both the nucleotide sequence and quality scores for each read. It is widely used in bioinformatics for a variety of applications, including genome assembly, read mapping, and variant calling.
SAM/BAM/CRAM
SAM, BAM, and CRAM are file formats used in bioinformatics for storing sequence alignment data. Here are their differences:
SAM (Sequence Alignment/Map) - This is a text-based format that contains alignment information of sequencing reads to a reference genome. It has a header section that includes information about the reference genome, and the alignment section contains data for each mapped read. SAM files are human-readable and can be easily manipulated, but they can be large in size and slow to parse.
BAM (Binary Alignment/Map) - This is a binary version of the SAM format, and it is more compact and faster to parse. BAM files are generated by converting SAM files using a tool like SAMtools. The alignment data is stored in binary format, making the files smaller and faster to access. BAM files can be indexed to enable rapid random access to specific parts of the file.
CRAM (Compressed SAM) - This is a compressed version of the BAM format, and it uses reference-based compression to reduce file size. It was developed to address the increasing size of BAM files, which can be a challenge for storage and analysis. CRAM files are significantly smaller than BAM files, and they can be decompressed to BAM format for compatibility with existing tools.
The main differences between these file formats are their file size, speed of access, and compression. SAM files are human-readable but large and slow to parse. BAM files are binary and more compact, but they still require substantial storage space and can be slow to access. CRAM files are compressed and even smaller than BAM files, but they require more computational resources to decompress, making them less suitable for quick random access.
Here's an example SAM file:
@HD VN:1.6 SO:coordinate
@SQ SN:chr1 LN:248956422
@SQ SN:chr2 LN:242193529
@SQ SN:chr3 LN:198295559
@PG ID:bwa PN:bwa VN:0.7.17-r1188 CL:bwa mem -t 8 ref.fa read1.fq read2.fq
read1 99 chr1 3052 60 76M = 3155 179 TGGTTAGGGTGTGTTTATGTTTTCAGGAGAAGATCTGTTAAGGTTAGTATCCAGTGAATCGTGGTATCGCGTCCCGCGCG CCCCCGGGGGGIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII X0:i:1 X1:i:0 MD:Z:76 NM:i:0
read2 147 chr1 3155 60 76M = 3052 -179 ACGTCATAGCTGCCACAGTTGCAATGACACGTTCTGGGTTAGGGACGAGTTCTGGGGGTGTTAGGGTGTGTTTATGTTTTCAG GGGGGIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII X0:i:1 X1:i:0 MD:Z:76 NM:i:0
In this example, the SAM file starts with header lines starting with the "@HD" and "@SQ" tags. These lines contain information about the reference genome used in the alignment and any other relevant information about the alignment process.
Following the header lines are alignment records, each consisting of a tab-separated line with 11 fields. Here's an explanation of the fields for each alignment record in the example:
| Field | Description |
| read1 | Read name |
| 99 | Flags indicating properties of the read and alignment (see SAM format specification for details) |
| chr1 | Reference sequence name |
| 3052 | 1-based leftmost mapping position |
| 60 | Mapping quality score |
| 76M | CIGAR string, indicating how the read aligns to the reference |
| \= | Reference sequence name of the mate/next read |
| 3155 | 1-based position of the mate/next read |
| 179 | Distance between the ends of the read and its mate/next read |
| TGGTTAGGGTGTGTTTATGTT...TATCGCGTCCCGCGCG | Sequence |
| CCCCCGGGGGGIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIIII | ASCII-encoded base quality scores |
| X0:i:1 | Optional fields, in the format TAG:TYPE:VALUE, indicating any additional information about the read and/or alignment |
| X1:i:0 | Another optional field |
| MD:Z:76 | Yet another optional field |
Here is a table of the fields in a SAM (Sequence Alignment/Map) file format:
| Field | Description |
| QNAME | Query template NAME, typically the read ID |
| FLAG | Bitwise FLAG indicating various properties of the read and alignment |
| RNAME | Reference sequence NAME, where the alignment is mapped |
| POS | 1-based leftmost mapping POSition of the first base in the read |
| MAPQ | MAPping Quality, the Phred-scaled probability that the alignment is incorrect |
| CIGAR | Extended CIGAR string describing the alignment |
| RNEXT | Reference sequence name of the mate/next read |
| PNEXT | Position of the mate/next read |
| TLEN | Observed Template LENgth, the signed observed length of the template |
| SEQ | Segment SEQuence, the bases of the read |
| QUAL | ASCII of Phred-scaled base QUALity+33 scores for each base in SEQ |
The optional fields, which are not required in all SAM files, provide additional information about the alignment and can be included by adding a tag and a value separated by a colon (:). The tag and value are separated by a tab. Some commonly used optional fields are:
| Field | Description |
| AS | Alignment Score, the score of the best alignment found |
| NM | Edit distance to the reference, the number of mismatches and gaps in the alignment |
| MD | String for mismatching positions, indicating the positions in the read that differ from the reference |
| RG | Read Group, identifying the group the read belongs to |
| SA | Supplementary Alignment, additional hits for the read |
The CIGAR string is a compact representation of the alignment between the query sequence and the reference sequence. It consists of a series of operators and numbers that indicate which parts of the query sequence aligned with the reference sequence and which parts do not. This provides a concise way of representing the alignment without writing out the entire sequence. The operators are defined in a table and are used in combination with numbers to indicate the length of the corresponding operation.
Here is a table of CIGAR operators and their corresponding descriptions:
| Operator | Description |
| M | Alignment match (can be a sequence match or mismatch) |
| I | Insertion to the reference |
| D | Deletion from the reference |
| N | Skipped region from the reference |
| S | Soft clipping (clipped sequences present in SEQ) |
| H | Hard clipping (clipped sequences NOT present in SEQ) |
| P | Padding (silent deletion from padded reference) |
| \= | Sequence match |
| X | Sequence mismatch |
Each operator is followed by a number that indicates the length of the corresponding operation. For example, "10M" indicates 10 bases that match the reference, while "3I" indicates 3 insertions to the reference.
In summary, SAM, BAM, and CRAM are all file formats used for storing sequence alignment data, with BAM being the binary version of SAM and CRAM being a compressed version of BAM. SAM files are human-readable and slow to parse, BAM files are binary and more compact but still require substantial storage space, and CRAM files are compressed and even smaller but require more computational resources to decompress. The choice of file format depends on the specific use case and the trade-off between file size, speed of access, and compression.
GVCF/VCF
GVCF (Genomic Variant Call Format) and VCF (Variant Call Format) are file formats commonly used to store information about genetic variants. GVCF is a special case of VCF that allows for the representation of incomplete or uncertain genotypes, while VCF is used to represent complete genotypes.
GVCF files differ from VCF files in that they contain information for each position in the genome, regardless of whether there is a variant present or not. This is accomplished by using a "reference block" to represent stretches of the genome where no variants are present, and by encoding incomplete or uncertain genotypes as either "no call" or a range of possible genotype calls using a special notation (e.g., "<MIN_DP>,<MIN_GQ>,<HOM_REF>,<NON_REF>"). This allows for efficient storage and analysis of large-scale variant data, such as those generated by whole-genome sequencing.
Here is an example of a VCF file:
##fileformat=VCFv4.3
##fileDate=2022-03-12
##reference=GRCh38.p13
##contig=<ID=1,length=248956422>
##contig=<ID=2,length=242193529>
##INFO=<ID=AF,Number=A,Type=Float,Description="Allele frequency">
##INFO=<ID=AC,Number=A,Type=Integer,Description="Allele count in genotypes">
##INFO=<ID=AN,Number=1,Type=Integer,Description="Total number of alleles in called genotypes">
##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype">
##FORMAT=<ID=DP,Number=1,Type=Integer,Description="Read Depth">
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT Sample1
1 10211 . A T 100 PASS AF=0.005;AC=10;AN=2000 GT:DP 0/0:42
1 10215 rs1234 G C 100 PASS AF=0.15;AC=300;AN=2000 GT:DP 0/1:60
2 20135 . C G,T 99 PASS AF=0.02,0.03;AC=4,6;AN=400 GT:DP 1/2:70
2 20987 . T C 50 LowQual AF=0.5;AC=1000;AN=2000 GT:DP 1/1:30
In the example file, the INFO field contains three key-value pairs describing allele frequency (AF), allele count (AC), and total number of alleles (AN) in the sample. The FORMAT field describes two fields for the sample data: genotype (GT) and read depth (DP). The sample data for each variant is listed in the corresponding row, with the genotype and read depth for "Sample1" listed in the last field.
Here is a table of the fields in a typical VCF file and their descriptions:
| Field | Description |
| #CHROM | Chromosome or contig name |
| POS | Position on the chromosome or contig |
| ID | Unique identifier for the variant |
| REF | Reference allele(s) |
| ALT | Alternate allele(s) |
| QUAL | Phred-scaled quality score of the evidence supporting the variant call |
| FILTER | Filter status (PASS, failed filters, or “.” for missing) |
| INFO | Additional information about the variant (e.g., allele frequency, functional annotations) |
| FORMAT | Description of the format of the sample genotype data |
| Sample(s) | Genotype information for each sample |
Here is a table of the additional fields in a typical GVCF file and their descriptions:
| Field | Description |
| #CHROM | Chromosome or contig name |
| POS | Position on the chromosome or contig |
| ID | Unique identifier for the variant |
| REF | Reference allele(s) |
| ALT | Alternate allele(s) |
| QUAL | Phred-scaled quality score of the evidence supporting the variant call |
| FILTER | Filter status (PASS, failed filters, or “.” for missing) |
| INFO | Additional information about the variant (e.g., allele frequency, functional annotations) |
| FORMAT | Description of the format of the sample genotype data |
| Sample(s) | Genotype information for each sample |
| GT | Genotype |
| GQ | Genotype quality |
| DP | Total depth |
| AD | Allelic depth |
| VAF | Variant allele frequency |
| PL | Phred-scaled genotype likelihoods |
| SB | Strand bias |
BED
The BED (Browser Extensible Data) format is a text file format used for storing genomic regions along with their associated annotations as coordinates. The data is organized in columns that are separated by spaces or tabs. Although this format was developed during the Human Genome Project and then adopted by other sequencing projects, its widespread use in bioinformatics had already made it a de facto standard before a formal specification was written.
Here's an example of a BED file:
chr1 100 500 my_feature 0 + 150 400 255,0,0 3 50,100,150 0,200,300
chr2 300 800 another_feature 0 - 400 700 0,255,0 4 50,75,125,150 0,200,500,650
One of the major benefits of the BED format is that it allows for the manipulation of coordinates instead of nucleotide sequences, which optimizes computation time when comparing all or part of genomes. Additionally, its simplicity makes it easy to parse and manipulate coordinates or annotations using word processing and scripting languages such as Perl, Python, or Ruby, or more specialized tools like BEDTools. Below is a table outlining the contents of a BED file:
| Column | Name | Description |
| 1 | Chromosome | Name of the chromosome where the feature is located |
| 2 | Start | 0-based starting position of the feature |
| 3 | End | 1-based ending position of the feature |
| 4 | Name | Name or identifier for the feature |
| 5 | Score | A score for the feature |
| 6 | Strand | The strand of the feature (+ or -) |
| 7 | ThickStart | Start position of the feature in a thick region |
| 8 | ThickEnd | End position of the feature in a thick region |
| 9 | ItemRGB | RGB color value for the feature |
| 10 | BlockCount | Number of blocks or exons in the feature |
| 11 | BlockSizes | Comma-separated list of block sizes |
| 12 | BlockStarts | Comma-separated list of block starting positions relative to the start position of the feature |
GTF/GFF
The GTF (Gene Transfer Format) and GFF (General Feature Format) file formats are used to describe genomic features such as genes, transcripts, and their attributes. Both formats are tab-separated text files, but the GFF format is more general and can describe any feature, while the GTF format is more specific and is typically used to describe gene and transcript models.
Example GTF file:
##gtf
chr1 hg19_refGene exon 11874 12227 . + . gene_id "NR_046018"; transcript_id "NR_046018"; exon_number "1"; gene_name "DQ598994"; gene_source "refgene"; gene_biotype "transcribed_unprocessed_pseudogene"; transcript_name "DQ598994"; transcript_source "refgene"; transcript_biotype "transcribed_unprocessed_pseudogene";
chr1 hg19_refGene exon 12613 12721 . + . gene_id "NR_024540"; transcript_id "NR_024540"; exon_number "1"; gene_name "DQ599062"; gene_source "refgene"; gene_biotype "transcribed_unprocessed_pseudogene"; transcript_name "DQ599062"; transcript_source "refgene"; transcript_biotype "transcribed_unprocessed_pseudogene";
chr1 hg19_refGene exon 13221 14409 . + . gene_id "NR_047525"; transcript_id "NR_047525"; exon_number "1"; gene_name "DQ599197"; gene_source "refgene"; gene_biotype "transcribed_unprocessed_pseudogene"; transcript_name "DQ599197"; transcript_source "refgene"; transcript_biotype "transcribed_unprocessed_pseudogene";
Example GFF file:
##gff-version 3
chr1 hg19_refGene exon 11874 12227 . + . gene_id=NR_046018; transcript_id=NR_046018; exon_number=1; gene_name=DQ598994; gene_source=refgene; gene_biotype=transcribed_unprocessed_pseudogene; transcript_name=DQ598994; transcript_source=refgene; transcript_biotype=transcribed_unprocessed_pseudogene;
chr1 hg19_refGene exon 12613 12721 . + . gene_id=NR_024540; transcript_id=NR_024540; exon_number=1; gene_name=DQ599062; gene_source=refgene; gene_biotype=transcribed_unprocessed_pseudogene; transcript_name=DQ599062; transcript_source=refgene; transcript_biotype=transcribed_unprocessed_pseudogene;
chr1 hg19_refGene exon 13221 14409 . + . gene_id=NR_047525; transcript_id=NR_047525; exon_number=1; gene_name=DQ599197; gene_source=refgene; gene_biotype=transcribed_unprocessed_pseudogene; transcript_name=DQ599197; transcript_source=refgene; transcript_biotype=transcribed_unprocessed_pseudogene;
Note that both the GTF and GFF files are in the same format, with the only difference being the separator character used to separate fields. In GTF files, fields are separated by tabs, while in GFF files, fields are separated by semicolons followed by a space character.
Here is a table describing the contents of a GTF and GFF file:
| Field | Description |
| seqname | The name of the sequence or chromosome where the feature is located. |
| source | The source of the feature, such as a program or database that generated it. |
| feature | The type of feature, such as gene, transcript, exon, or CDS. |
| start | The starting position of the feature, 0-based inclusive. |
| end | The ending position of the feature, 0-based exclusive. |
| score | The score of the feature, usually a floating-point value. |
| strand | The strand of the feature, either '+' or '-'. |
| frame | For features that have a reading frame, such as CDS or start/stop codons, this field indicates the offset of the first complete codon within the feature. |
| attributes | A semicolon-separated list of attribute-value pairs that describe the feature. The attributes can include gene ID, transcript ID, gene name, gene biotype, exon number, and other annotations. The attribute "gene_id" is typically used to link together all of the features that belong to the same gene. |
Some differences between GTF and GFF format:
GTF is more specific to gene and transcript models while GFF is more general and can describe any feature.
GFF is a more flexible format that allows users to define their own attributes, whereas GTF has a fixed set of attributes.
The attribute field in GFF format can have an arbitrary number of key-value pairs, while the attribute field in GTF format is typically limited to a few well-defined keys.
Overall, both GTF and GFF are widely used file formats for describing genomic features and their attributes. They can be used to store gene and transcript models, as well as other genomic features such as regulatory regions, repeat elements, and chromatin domains.
GBK/GBF
The GenBank (GBK) and GenBank Flat File (GBF) formats are file formats commonly used to store and exchange genomic data. Both formats contain annotations and metadata for genomic sequences, including genes, coding regions, and other features. The main difference between the two formats is that GBF files are flat files with one record per sequence, whereas GBK files can contain multiple records for a single sequence.
Here are some example files in both GBK and GBF formats:
Example GBK file:
LOCUS NC_000913 4641652 bp DNA circular BCT 15-JUL-2021
DEFINITION Escherichia coli str. K-12 substr. MG1655, complete genome.
ACCESSION NC_000913
VERSION NC_000913.3
DBLINK BioProject: PRJNA577777
Assembly: GCF_000005845.2
GenBank: NC_000913.3
FEATURES Location/Qualifiers
source 1..4641652
/organism="Escherichia coli str. K-12 substr. MG1655"
/mol_type="genomic DNA"
/strain="K-12 substr. MG1655"
/db_xref="taxon:511145"
gene complement(190..255)
/locus_tag="b0001"
/old_locus_tag="ECK0001"
/gene="thrA"
/note="thr operon leader peptide"
/codon_start=1
/transl_table=11
/product="aspartokinase"
/protein_id="NP_414542.1"
/db_xref="ASAP:ABE-0000006"
/db_xref="UniProtKB/Swiss-Prot:P00509"
/db_xref="EcoGene:EG11277"
/db_xref="GeneID:944742"
CDS complement(190..255)
/locus_tag="b0001"
/old_locus_tag="ECK0001"
/gene="thrA"
/note="thr operon leader peptide"
/codon_start=1
/transl_table=11
/product="aspartokinase"
/protein_id="NP_414542.1"
/db_xref="ASAP:ABE-0000006"
/db_xref="UniProtKB/Swiss-Prot:P00509"
/db_xref="EcoGene:EG11277"
/db_xref="GeneID:944742"
/translation="MTAEELDSLFDRTLREIGDALARTIDVKVGEVAVGITAVVAQALK
DQGKRVVLLSHGAVLHPNIDQIQLLDLSDGEILFVAPSTGEVLPALKTFVQALQRG
RPLIAIVNAGSGMNGVHVKIAAPGQIAQAIANEVLASGGNNIAVIAAEPGSIENVIG
MTTAAGHPMRSVADVIAAGTPIAVEVFTGLSRLENPKLDAGTAHGGHLTHGAFIALD
EPTAPLAGFAAVNKISLAEQVRTISAEVHGRVRTGRWEKAEVRAALVDGNAVVVLDG
GTPTQQIAEALALMGASAIAGVAHPMIVPVKDLTDPAATLEALAEKLNLPAMEVQRL
AGQEGDELYKLCDEILGLLRSLVEYLAPGAFTLPPAQIAQAAGIPLPVVNFKESPEQA
VREVLQHGILDSVIASVGRAVRQTALRDLMFSLAKEGTPLLLSGVVGFLEPFTLDTL
FRTEDLSGQIQRVGLRAYSLGVNLPVWRTARPEE
Here is a table of some of the common fields found in both formats:
| Field | Description |
| LOCUS | Information about the sequence, such as its name and length |
| DEFINITION | A brief description of the sequence and its features |
| ACCESSION | The unique identifier for the sequence |
| VERSION | The version of the sequence |
| KEYWORDS | Keywords that describe the sequence or its features |
| SOURCE | Information about the source organism or sample from which the sequence was obtained |
| FEATURES | Annotations for the sequence, including genes, exons, and other features |
| ORIGIN | The actual DNA or RNA sequence |
NGS Data Formats Summary
| Data Format | Description | Data Content |
| FASTA | Text-based format containing nucleotide or amino acid sequences | DNA or protein sequences |
| BCL | Binary base call format generated by Illumina sequencing machines | Raw sequencing data, quality scores, signal intensity |
| FASTQ | Text-based format containing nucleotide sequences and their corresponding quality scores | Raw sequencing reads |
| BAM | The binary version of the SAM format containing aligned reads and associated metadata | Aligned sequencing reads, quality scores, alignment information |
| CRAM | A compressed version of the BAM format, reducing storage requirements while retaining full data fidelity | Aligned sequencing reads, quality scores, alignment information |
| VCF | Variant Call Format used for storing genomic variations such as SNPs, indels, and structural variants | Genomic variations, genotypes, quality scores |
| BED | Browser Extensible Data format used for storing genomic features such as genes, exons, and regulatory elements | Genomic feature coordinates |
| GTF/GFF | Gene Transfer Format/General Feature Format used for storing gene annotations and other genomic features | Gene annotations, genomic feature coordinates |
| GBK/GBF | The file format used for storing annotated DNA and protein sequences | DNA or protein sequences, annotations, features, references |
Workflow Managers in NGS Data Analysis
Workflow managers play a crucial role in Next-Generation Sequencing (NGS) data analysis by automating the processing of large datasets and ensuring the reproducibility of results.
Here are some of the benefits of using popular workflow managers:
Automation of data processing: NGS data analysis involves several steps, from data preprocessing to downstream analysis. Workflow managers automate this process, saving time and minimizing human error.
Reproducibility: Workflow managers ensure that the same analysis pipeline is run with the same input data, parameters, and tools, producing reproducible results. This is especially important in research settings where reproducibility is essential for scientific validity.
Scalability: Workflow managers can handle large datasets with ease and can be run on a variety of platforms, including local machines, clusters, and cloud computing services.
Portability: Workflow managers allow users to write workflows in a language-agnostic manner, making them portable across different platforms and environments.
Modularity: Workflow managers allow users to modularize complex analysis pipelines into reusable components, simplifying workflow design and maintenance.
Snakemake, Nextflow, Cromwell, and Galaxy are all popular workflow managers used in NGS data analysis. Snakemake is a Python-based workflow management system, Nextflow is a polyglot workflow management system that supports multiple languages, Cromwell is a workflow management system that integrates with Google Cloud Platform, and Galaxy is a web-based platform that provides a graphical interface for workflow design and analysis.
Overall, workflow managers are essential for efficient, reproducible, and scalable NGS data analysis.
Snakemake is a Python-based workflow management system that simplifies the creation and execution of complex workflows. It uses a domain-specific language that allows users to define the input, output, and steps required to complete a specific analysis. Snakemake's strength lies in its ability to handle large and complex datasets and parallelize computational tasks to optimize resource utilization.
Nextflow is a workflow management system designed to simplify the creation and execution of reproducible and scalable workflows. It is built using a domain-specific language that is agnostic to the underlying computing infrastructure, allowing for seamless deployment on local machines, clusters, and cloud platforms. Nextflow's robust error handling and built-in support for containerization make it a powerful tool for managing complex data analysis pipelines.
Cromwell is an open-source workflow management system designed to simplify the creation, execution, and monitoring of workflows on a variety of computing infrastructures. It provides a powerful and flexible way to manage complex data analysis pipelines using a declarative language that allows users to define the inputs, outputs, and dependencies of their workflows. Cromwell is widely used in the scientific community for its support of multiple workflow languages, built-in error handling, and extensive logging and monitoring capabilities.
Galaxy is a web-based platform that enables users to create, run, and share reproducible data analysis workflows. It provides a user-friendly interface that allows researchers to access a wide range of bioinformatics tools and pipelines without requiring advanced programming skills. Galaxy's strength lies in its ability to integrate with a variety of data sources, tools, and pipelines, making it a powerful tool for both novice and experienced bioinformaticians.
Here's a table comparing the four popular workflow managers for NGS data analysis, along with their initial release date and programming language:
| Workflow Manager | Initial Release Date | Programming Language |
| Snakemake | 2012 | Python |
| Nextflow | 2013 | Groovy/Java |
| Cromwell | 2016 | Java/Scala |
| Galaxy | 2005 | Python/JavaScript |
In the coming articles, we will focus on a comparison between two popular workflow management systems, Snakemake and Nextflow.
Snakemake V/S Nextflow
Snakemake and Nextflow are both workflow management systems designed to simplify the management of complex scientific workflows. Both tools enable the reproducibility of analyses and are capable of executing tasks in parallel, making them particularly useful for large-scale data analysis.
One of the main differences between Snakemake and Nextflow is the language used to write the workflow scripts. Snakemake uses a Python-based domain-specific language (DSL), whereas Nextflow uses a Groovy-based DSL. This means that users familiar with Python may find Snakemake more intuitive to use, while those more comfortable with Groovy may prefer Nextflow.
Another difference is in the way that the tools handle dependencies. Snakemake has built-in support for conda, which enables users to create isolated software environments with specific dependencies. Nextflow also supports conda, but also has support for Docker and Singularity containers, making it easier to manage dependencies across different computing environments.
Finally, there are differences in the way that the tools handle error reporting and recovery. Snakemake provides a more detailed error report than Nextflow, making it easier to identify and debug issues in the workflow. However, Nextflow has a more robust error recovery mechanism, which enables workflows to resume from the point of failure, rather than having to start from scratch.
Overall, both Snakemake and Nextflow are powerful tools for managing complex scientific workflows, and the choice between them will depend on the specific needs and preferences of the user.
Snakemake Example
Here is an example Snakemake workflow code for running FastQC and MultiQC:
# Define input and output files
fastq_dir = "data/fastq"
sample = ["A1","B1"]
results_dir = "results"
# Define rule for running FastQC
rule fastqc:
input:
expand(f"{fastq_dir}/{sample}.fastq.gz", sample=sample),
output:
directory(f"{results_dir}/fastqc/{sample}")
container:
"fastqc:latest"
shell:
"fastqc {input} --outdir {output}"
# Define rule for running MultiQC
rule multiqc:
input:
expand(f"{results_dir}/fastqc/{{sample}}", sample=sample),
output:
directory(f"{results_dir}/multiqc")
container:
"multiqc:latest"
shell:
"multiqc {input} --outdir {output}"
# Define the workflow
rule all:
input:
expand(f"{results_dir}/multiqc", sample=sample)
The workflow starts by defining the input and output files. The input files are located in data/fastq and are in the format of {sample}.fastq.gz. The output files will be located in results.
The first rule (fastqc) defines the input and output files for running FastQC on each input file. It specifies the Docker container to use (fastqc:latest) and runs the fastqc command on the input file to generate a report in the output directory.
The second rule (multiqc) defines the input and output files for running MultiQC on all of the FastQC reports generated by the previous rule. It specifies the Docker container to use (multiqc:latest) and runs the multiqc command on all of the FastQC reports to generate a summary report in the output directory.
Finally, the workflow specifies that the output files for the multiqc rule should be used as the final output for the workflow. This is done with the rule all statement, which takes the form of input: [...] and specifies the final output files. In this case, it uses the expand function to generate a list of all of the multiqc output directories (one for each input file).
Note: This code assumes that the Docker containers for FastQC and MultiQC are already installed on the system. If they are not, they can be downloaded and installed using the docker pull command.
Nextflow Example
Here's an example Nextflow workflow code for running FastQC and MultiQC on multiple input FASTQ files:
// Define input files
params.reads = Channel.fromPath("data/*.fastq.gz")
// Define FastQC process
process fastqc {
container 'fastqc:latest'
input:
file reads from params.reads
output:
file "*.zip" into qc_reports
script:
"""
fastqc -o ${qc_reports} ${reads}
"""
}
// Define MultiQC process
process multiqc {
container 'multiqc:latest'
input:
file reports from qc_reports.collect()
output:
file "multiqc_report.html" into final_report
script:
"""
multiqc -o ${final_report} ${reports}
"""
}
// Define output location
output:
file "multiqc_report.html" into "results"
// Run the workflow
workflow {
fastqc
multiqc
}
This Nextflow script takes as input a set of FASTQ files (in this case, all files in the data/ directory with a .fastq.gz extension), runs FastQC on each file to generate QC reports and then runs MultiQC to aggregate the results of all FastQC reports into a single HTML report. The workflow is containerized using Docker images for FastQC and MultiQC, which are specified with the container directive.
The script defines two processes: fastqc and multiqc. The fastqc process takes one input file (a single FASTQ file) and produces one output file (a ZIP archive containing FastQC results). The multiqc process takes all FastQC results generated by the fastqc process as input and produces a single HTML report as output. The collect() method is used to gather all FastQC reports generated by the fastqc process.
The output location is defined to save the final MultiQC report in a results directory. The workflow block specifies the order of execution for the two processes.
Note that this is a simplified example and a real-world workflow might have additional processing steps, more complex input/output specifications, and use more advanced features of Nextflow such as channel broadcasting, error handling, and caching.
I hope you found this article informative and gained some knowledge about NGS data formats and workflow managers. In the upcoming articles, we will explore how to create a complete workflow using Nextflow and Snakemake. Thank you for reading!
To gain a general understanding of next-generation sequencing (NGS) or to refresh your knowledge on the topic, please refer to the following article:
For more information on sequence cleaning and quality control of NGS data, please refer to the following article:
Subscribe to my newsletter
Read articles from Bhagesh Hunakunti directly inside your inbox. Subscribe to the newsletter, and don't miss out.
Written by
Bhagesh Hunakunti
Bhagesh Hunakunti
I'm a science guy with a creative instinct. Simple-minded & doing what I'm good at & sharing what I've learnt so far with amazing people like you'll.