BAM Files: When Sequence Reads Match with References
 Lucas Taniguti
Lucas Taniguti
No matter if you obtained your sequence from Illumina, Nanopore or Sanger, it's always possible to align their nucleotide bases against a reference sequence. It is common to call this reference sequence a "Reference Genome" if it represents the whole DNA sequence of a species and is broadly used in bioinformatics. For example Human Genome or Ustilago maydis (fungus)
In this article, I'll quickly explain the alignment process, the kind of information BAM files contain and why they are important for bioinformatics. Hope you enjoy it!
Where do BAM files come from?
(Quick answer: alignments)
When working with Next-Generation Sequencing (NGS) it is common to have millions of sequence reads, stored usually in pairs of FASTQ files. At this point, you probably know nothing about the content of your sequencing experiment. I mean, you just have gigabytes of data in computer files with those A, C, T and Gs.
I've seen some anxious people simply opening one of these FASTQ files, copying the content of one read and using BLASTn to know if the data they are receiving have come from the expected species. While it can be helpful for quick answers it's not the right way.
Usually, you know the species you have sequenced and probably there is a reference sequence available. Then it's very straight forward perform the alignment.
In this post, I'll not dive into de novo genome assembly challenges and metagenomics experiments.
The alignment process is often compared to a jigsaw puzzle, where each piece is a fragment of DNA (represented in your FASTQ files), and the picture on the box is the reference sequence. In essence, alignment is the mechanism of fitting these pieces together in a manner that aligns with the reference picture, thereby illuminating the genetic architecture of an organism.

For a clear understanding of their origin, let's explore a straightforward example. The following code demonstrates how we can achieve an alignment from paired-end sequencing data using BWA-MEM:
bwa mem refs/Homo_sapiens_assembly38.fasta \
reads/NA12878.pe.R1.fastq.gz \
reads/NA12878.pe.R2.fastq.gz > alignment.sam
Let's break it down:
bwa mem: This invokes the BWA-MEM algorithm from the BWA software package. BWA-MEM is widely used for aligning sequencing reads to a large reference genome.refs/Homo_sapiens_assembly38.fasta: This specifies the path to the reference genome that the sequencing reads will be aligned against. In this case, it's a FASTA file containing the human reference genome (GRCh38/hg38).reads/NA12878.pe.R1.fastq.gzandreads/NA12878.pe.R2.fastq.gz: These are the paths to the input sequencing read files. In this case, these are paired-end reads (R1andR2) from the sample NA12878. They are in FASTQ format and have been compressed using gzip (hence the.gzextension).> alignment.sam: This part of the command line is redirecting the output of the BWA-MEM command to a file namedalignment.sam. The>symbol in a command line means "redirect the output to a file." The output file here is in SAM format.
Overall, this command is aligning the sequencing reads in the specified FASTQ files to the human reference genome using BWA-MEM, and the resulting alignments are being saved in a SAM format file named alignment.sam.
What BAM files are?
When you learn about BAM files you'll also be learning about SAM and CRAM. These two other formats have the same information, only the compression strategy changes.
SAM files (Sequence Alignment Map) are just text files, without any compression
BAM files (Binary Alignment Map) are compressed using BGZF (Blocked GNU Zip Format). Similar to the GZIP format, you probably already had experience using UNIX systems
CRAM files (Compressed Reference-oriented Alignment Map) only store the differences between the read and the reference, which requires significantly less space. This is particularly effective when dealing with high-coverage datasets where much of the information is duplicated across reads. It heavily relies on the reference sequence during the compression and decompression process.
You will often see a companion index for BAM and CRAM files. Similar to .fai we already saw in the FASTA files, it enables programs to quickly access particular sections of the file, eliminating the need to read it in its entirety. Its extensions are .bai and .crai, respectively.
From here on I'll use "alignment file" when the compression level is not relevant.
Each sequencing read needs to have information about where is its best match against the reference sequence and how it is aligned. This is the core information in an alignment file.
If you're interested in exploring every intricate detail of the data within an alignment file, please refer to the official PDF document titled 'Sequence Alignment/Map Format Specification'. It provides an exhaustive explanation of the format's specifications, ensuring a comprehensive understanding.
The file comprises a header section (denoted by lines beginning with '@') and individual lines depicting the alignment of each read derived from your FASTQ files. Here's an illustrative example representing a paired alignment of a single DNA fragment (reads originating from the same DNA fragment, found in R1 and R2 files):
@HD VN:1.6 SO:coordinate GO:query
@RG ID:A SM:NA12878
READ1 83 chrM 16097 60 100M = 15930 -268 <READ SEQ> <READ QUAL> <other metadata columns>
READ1 163 chrM 15930 60 100M = 16097 268 <READ SEQ> <READ QUAL> <other metadata columns>
In the image (of a table!) below you can see the mandatory columns of the alignment section. As you can imagine, because of the complexity, I just copy/paste from the official spec documentation.
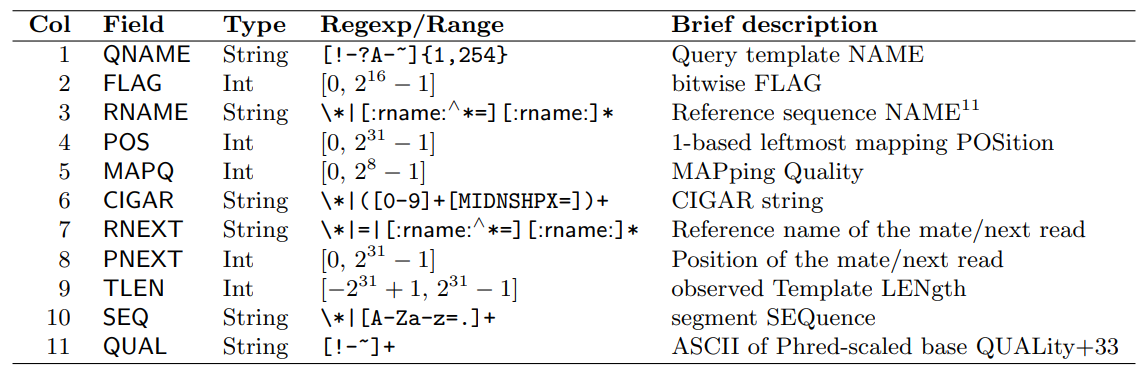
This mysterious bitwise FLAG is just telling you more information about the read. Check this tool Decoding SAM flags for better understanding.
🐘Note: when working with Broad's GATK you will need to inform the @RG header line.
What we can do with alignment files?
Alignment files have a lot of information inside, so there are a lot of things we can do with them. Here I point out some of these analyses.
Visualizing alignments: alignment files can be loaded into genome browsers, like UCSC Genome Browser or Integrative Genomics Viewer (IGV), for visual inspection. This allows researchers to manually examine how the reads align to the reference and look for regions of interest or unusual patterns.
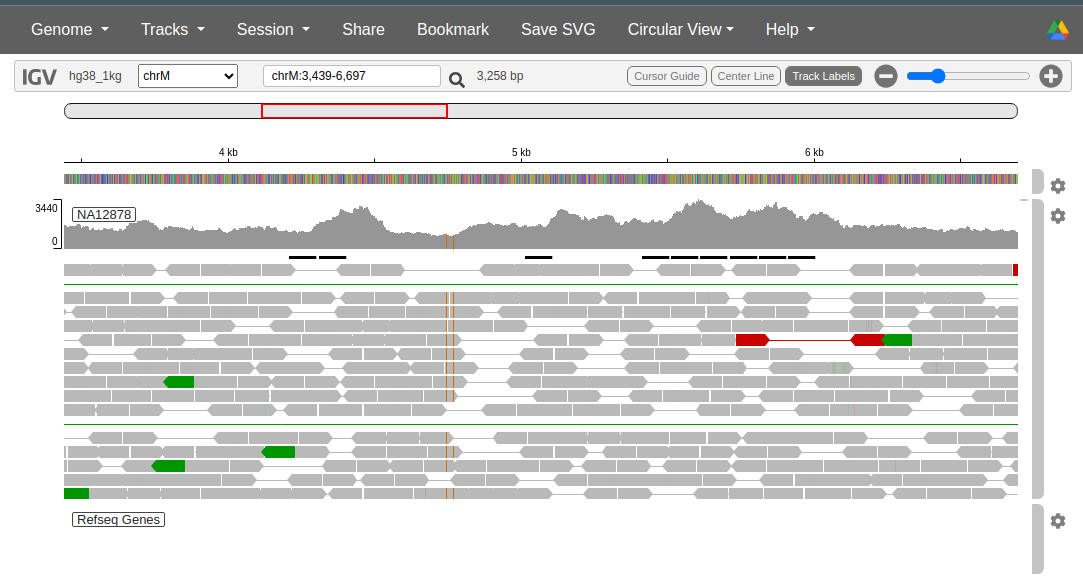
The figure shows an example of an alignment between paired-end Illumina reads and the reference genome, highlighting a region in the mitochondrion.
Variant calling: alignment files are typically used as input for variant calling tools like GATK, FreeBayes, or Samtools mpileup. These tools look for differences between the aligned reads and the reference sequence to identify genetic variants such as single nucleotide polymorphisms (SNPs) or insertions and deletions (indels).
🐘Note: Variants are typically stored in VCF files. If you're interested in understanding this format, make sure to subscribe! My next post will likely explore it.
Quantifying expression levels: In RNA-Seq analysis, they can be used to quantify transcript expression levels. Tools like HTSeq or featureCounts count the number of reads that align to each gene or transcript, which is used as a measure of expression level.
Identifying structural variants: like large insertions, deletions, duplications, inversions, or translocations. Tools for this task include DELLY, LUMPY, and Manta. I also find the material provided by the Broad Institute very instructive, which explains how to interpret structural variants based on the orientation of the aligned pairs. Check it out here!
Checking sequencing and alignment quality: alignment files contain a wealth of information about the quality of sequencing and alignment, including per-base quality scores, mapping quality scores, and flags indicating any issues with the alignment. This information can be used to troubleshoot issues with the sequencing run or the alignment process. You can use a tool like Mosdepth or Picard CollectAlignmentSummaryMetrics
Commonly used tools for doing alignments
Bellow I point out some software that can be used for the specific goals of aligning short/long reads from DNA/RNA sequencing experiments.
BWA-MEM
It is among the most popular aligners and probably will be the one you see more frequently, mainly because of the success of Illumina sequencing. It is part of the BWA software package, and I think it stands for the Burrows-Wheeler Aligner with Maximal Exact Matches. In the original paperwork, it's described as:
BWA-MEM follows the canonical seed-and-extend paradigm. It initially seeds an alignment with supermaximal exact matches (SMEMs) using an algorithm we found previously (Li, 2012, Algorithm 5), which essentially finds at each query position the longest exact match covering the position - PDF link
So, maybe it should be named BWA-SMEM?🤔. I'm not going to dig into it!
What is important is that it is a widely used software package for mapping low-divergent sequences against a large reference genome, such as the human genome. It works efficiently with reads from 70bp to a few megabases. By low-divergent sequences, you can think of one sequencing experiment with high base-call accuracy or aligning sequencing reads to a very closely related genome sequence.
STAR
Stands for Spliced Transcripts Alignment to a Reference, it is widely used for the alignment of RNA sequencing data. Its focus is on spliced alignments, a necessary feature for RNA-seq data. Unlike DNA, mature RNA often contains only exonic regions - introns are removed in a process called splicing. These spliced sequences need to be aligned back to the genomic sequence, which is a task that STAR excels in.
Minimap2
It is capable of aligning longer reads, even up to 1 million bp, making it particularly suitable for long-read sequencing technologies like those from Pacific Biosciences or Oxford Nanopore. In its official code repository (GitHub) they describe the main use cases of this algorithm. I'll just paste the content here:
Mapping PacBio or Oxford Nanopore genomic reads to the human genome;
Finding overlaps between long reads with error rates up to ~15%;
Splice-aware alignment of PacBio Iso-Seq or Nanopore cDNA or Direct RNA reads against a reference genome;
Aligning Illumina single- or paired-end reads;
Assembly-to-assembly alignment;
Full-genome alignment between two closely related species with divergence below ~15%.
In conclusion, the choice of aligner depends on your data and the nature of your experiment. For high-quality short reads from a DNA sequencing experiment, consider using BWA-MEM. If you're dealing with RNA sequencing, STAR is likely to be your best option. When working with long reads, which often exhibit higher sequencing error rates, Minimap2 is highly recommended.
There are a high number of other aligners out there. To name some of them: bowtie, tophat, bwa-mem2, LAST, HISAT2.
That's it for today. Our next steps will be exploring the files containing genetic variants: the VCF files! Feel free to share your thoughts or ask any questions in the comments below. Make sure to subscribe!
Subscribe to my newsletter
Read articles from Lucas Taniguti directly inside your inbox. Subscribe to the newsletter, and don't miss out.
Written by
Lucas Taniguti
Lucas Taniguti
I am a bioinformatician and backend developer at Mendelics, a leading genetic diagnostic company in Latin America.