Exploring Spider Genomes: Assessing Assembly Completeness
 Lucas Taniguti
Lucas Taniguti
When I was a child, my parents gifted me a fascinating magazine series dedicated to the discovery of Earth's "mini monsters" – tiny creatures that inhabit our planet. Little did I know that today dozens of spider genomes would be available, and scientists would be studying their genes. I read that especially those related to silk and venom production, to understand how these substances are produced and potentially harness them for various applications, such as in materials science or medicine.

Figure showing the assembled glow-in-the-dark scorpion and spider puzzles from the mini-monsters magazine collection (1994). Each magazine came with a portion of the puzzle pieces.
Enough with nostalgia! 😅
Today, we're going to learn about a new tool named 'compleasm'. This tool helps us check how good genome assemblies are. Even if you're new to this work, it can be handy. It's also a good way to see the quality of spider genomes we have now.
Note that I never worked with spider genomes. I'm using it here just to check the results of busco and compleasm.
Quality Analysis with BUSCO
By comparing a genome assembly against a predefined set of highly conserved single-copy orthologous genes, BUSCO provides a valuable measure of the assembly's completeness. It allows researchers to evaluate the extent to which the genome under analysis contains the expected genes.
The use of this tool has become an integral part of the bioinformatician's toolkit for genome assembly. Indeed, its adoption is so widespread that it is commonly used to assure readers of scientific papers that the genome assembly is complete, or at the very least, nearing completion.
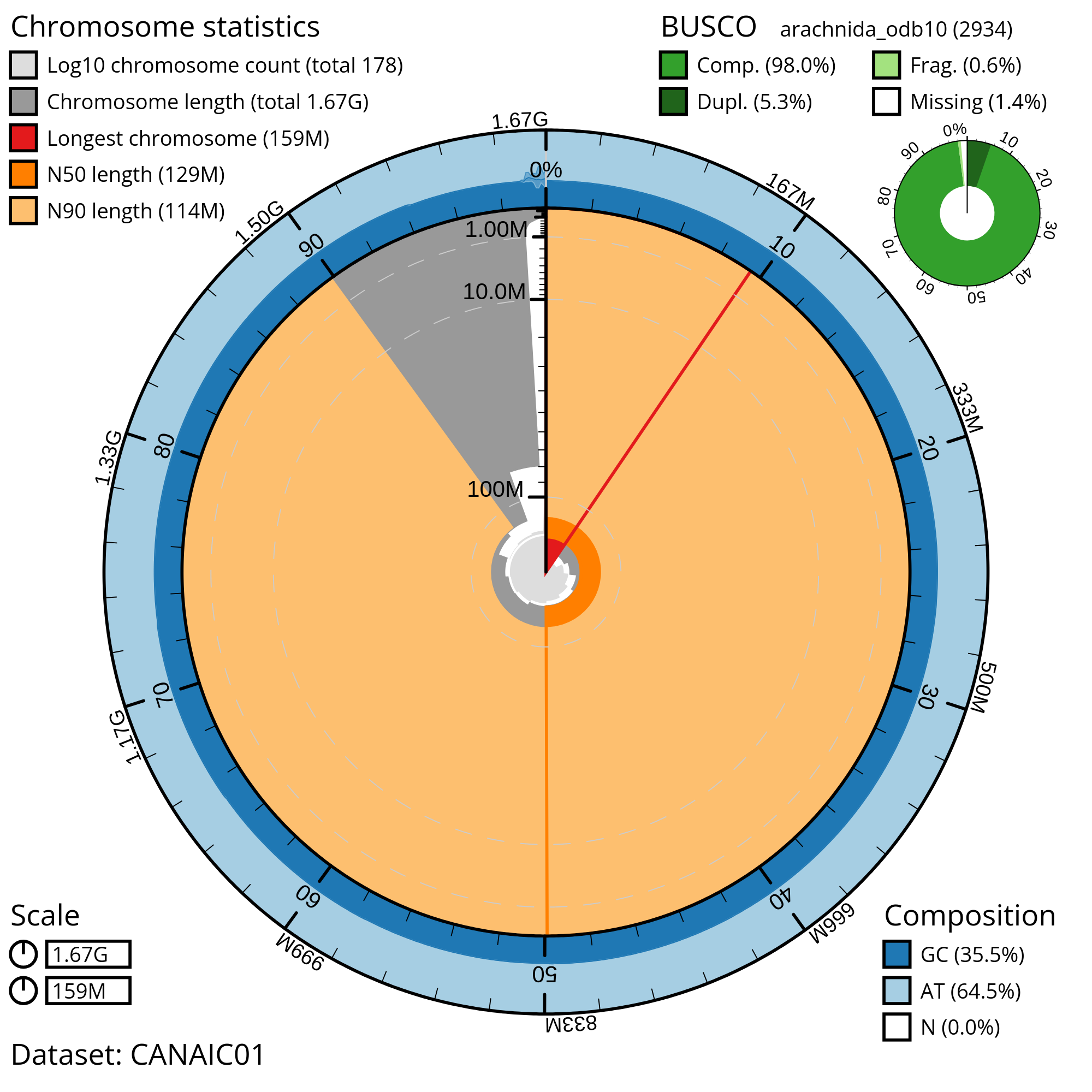
If you are interested in this topic check out this web browser tool that exhibits several metrics of published genomes: blobtoolkit.
Introducing the New Player
This year we had a new addition to the field of bioinformatics, with the introduction of compleasm, another tool designed for genome completeness analysis.
This seems to be a great tool, developed by Neng Huang and Heng Li. Heng Li contributions to the field include several useful other tools such as bwa, samtools, bcftools, and seqtk, which are commonly utilized by many bioinformaticians. Check out the pre-print of this work:
miniBUSCO: a faster and more accurate reimplementation of BUSCO. Neng Huang, Heng Li. bioRxiv 2023.06.03.543588; doi: https://doi.org/10.1101/2023.06.03.543588
It will probably have the name changed to compleasm in the paper final version.
The main advantages of compleasm are that (quoted from the article):
"achieves significant improvement over BUSCO by 3.4 to 14.5 times. Moreover, the speedup tends to increase with larger genome sizes"
"miniBUSCO reported completeness of 99.6% compared to 95.7% reported by BUSCO. Among them, 562 complete genes were only reported by miniBUSCO" ... "We found that 557 out of 562 miniBUSCO-specific complete genes could be supported by the annotation."
"Separating frameshift errors from assembly incompleteness is a unique advantage of miniBUSCO"
"suggests running BUSCO additionally to ensure a more reliable assessment of assembly completeness for input assemblies with high divergence"
In conclusion, compleasm presents a significant advancement for assembly evaluation. It states to outperform its predecessor, BUSCO, in terms of both efficiency and accuracy, particularly with larger genome sizes. It provides a more comprehensive and accurate assessment of genome completeness, identifying complete genes that BUSCO does not. Furthermore, its unique ability to separate frameshift errors from assembly incompleteness adds another layer of depth to its analysis. However, it is worth noting that for highly divergent assemblies, the use of BUSCO in conjunction with compleasm is recommended to ensure the most reliable assessment. As genome sequencing technologies continue to evolve, tools like compleasm will play an increasingly important role in ensuring the quality and completeness of genome assemblies.
Let's get our hands dirty
I understand that many of you appreciate hands-on practice, so in this post, we're going to reproduce a result presented in a recent paper - a scatter plot that correlates the BUSCO results with compleasm, focusing exclusively on spider genomes. The compute-intensive part of this analysis can be found in a gist, which will be linked at the end of this blog post.
Here is our plan:
Get some complete spider genomes from NCBI
Run compleasm for each one
Get BUSCO metrics from blobtoolkit
Prepare a table and a plot with the results
This plot summarizes our little hands-on:
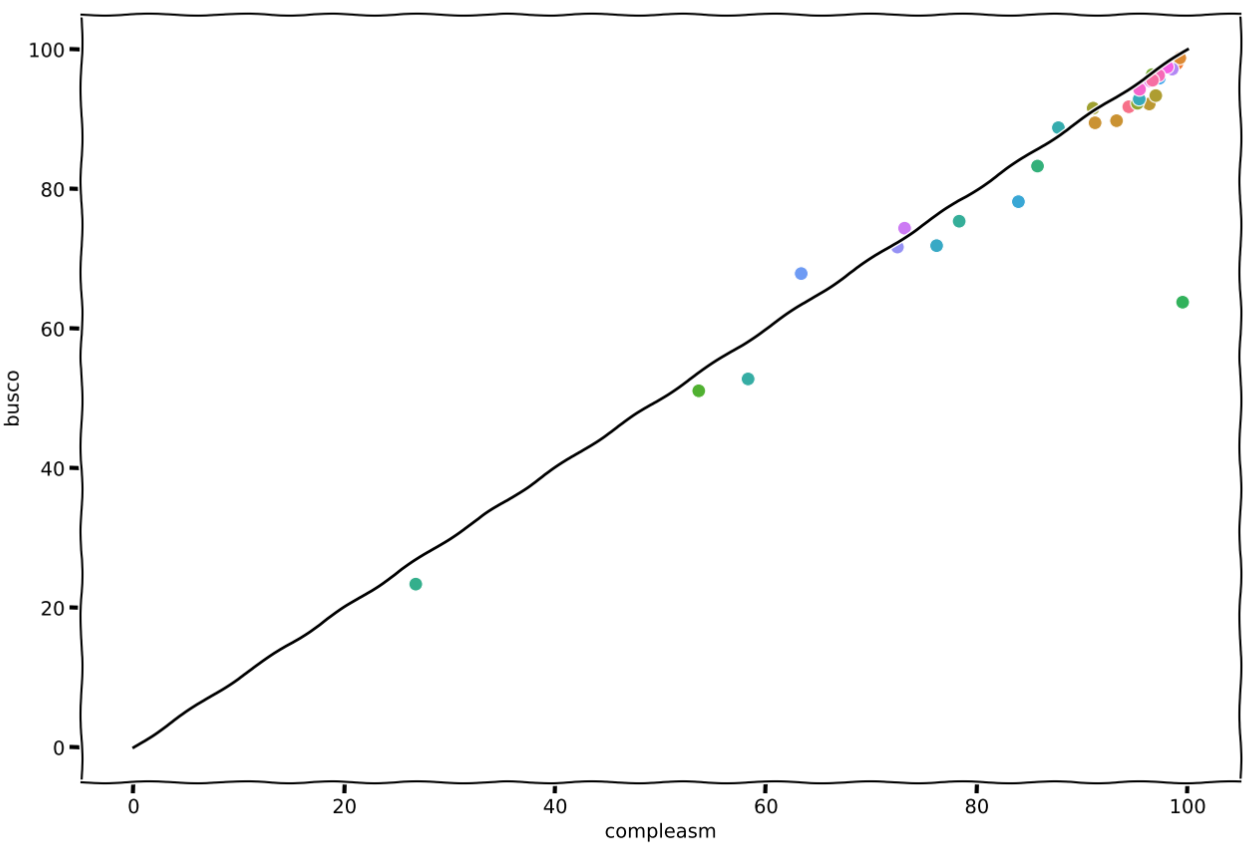
Scatter plot showing the numbers of completed genes reported by compleasm and BUSCO. We see here what authors already have described in their paper: "miniBUSCO (compleasm) reports a higher completeness range of 0-12% than BUSCO". The exception here is the assembly GCA_021527715.1, which went from 63.8% to 99.52% of completeness (diff of ~32%). I'll probably need to double check it🧐
Acquiring Spider Genomes
To circumvent the necessity of running BUSCO, I've opted for genomes with readily available metrics from the blobtoolkit.
Executing compleasm
A Docker image for this new tool is conveniently provided by the biocontainers community. For its execution, we will use this specific image:
quay.io/biocontainers/minibusco:0.2.1--pyh7cba7a3_0
For each spider genome under analysis, we will utilize the arachnida_odb10 BUSCO dataset to assess the completeness of the assembly.
Parsing output to make the plot
To make the plot we had to parse and tabulate all metrics and results. In short, we downloaded the table from blobtoolkit that summarizes the assemblies and then merged it into a pandas data frame with data from compleasm.
We use seaborn to craft the compelling scatterplot (using xkcd theme!). The table containing the data used for the plot is provided below.
Accession | Taxon | busco | compleasm |
GCA_021605095.1 | Caerostris extrusa | 91.8 | 94.41 |
GCA_907164885.1 | Dolomedes plantarius | 98 | 98.84 |
GCA_947359465.1 | Metellina segmentata | 98 | 98.98 |
GCA_949128135.1 | Parasteatoda lunata | 98.8 | 99.25 |
GCA_012066115.1 | Tyrophagus putrescentiae | 89.5 | 91.21 |
GCA_013358835.1 | Ixodes persulcatus | 92.2 | 96.35 |
GCA_013339685.1 | Hyalomma asiaticum | 93.4 | 96.97 |
GCA_013372475.1 | Leptotrombidium pallidum | 91.6 | 91.03 |
GCA_015342795.1 | Argiope bruennichi | 92.3 | 95.23 |
GCA_018873245.1 | Archegozetes longisetosus | 96.4 | 96.63 |
GCA_022884545.1 | Speleorchestes sp. AD1671 | 51.1 | 53.61 |
GCA_021730765.1 | Tyrophagus putrescentiae | 89.8 | 93.25 |
GCA_021527715.1 | Neoseiulus cucumeris | 63.8 | 99.52 |
GCA_000697925.2 | Latrodectus hesperus | 83.3 | 85.75 |
GCA_000973045.2 | Ixodes ricinus | 23.4 | 26.76 |
GCA_002135145.1 | Euroglyphus maynei | 75.4 | 78.32 |
GCA_000484575.1 | Mesobuthus martensii | 52.8 | 58.29 |
GCA_003675905.2 | Leptotrombidium deliense | 88.8 | 87.73 |
GCA_002943765.1 | Psoroptes ovis | 92.9 | 95.4 |
GCA_003123905.1 | Cordylochernes scorpioides | 71.9 | 76.18 |
GCA_006491805.2 | Dysdera silvatica | 78.2 | 83.94 |
GCA_008065355.1 | Pardosa pseudoannulata | 95.9 | 97.31 |
GCA_015350385.1 | Aculops lycopersici | 67.9 | 63.33 |
GCA_011317285.1 | Androctonus mauritanicus | 71.7 | 72.46 |
GCA_000611955.2 | Stegodyphus mimosarum | 97.2 | 98.53 |
GCA_023170135.1 | Acarapis woodi | 74.4 | 73.14 |
GCA_019973975.1 | Trichonephila clavata | 95.5 | 96.45 |
GCA_019973935.1 | Trichonephila clavipes | 97.5 | 98.06 |
GCA_019973955.1 | Trichonephila inaurata madagascariensis | 94.3 | 95.43 |
GCA_019974015.1 | Nephila pilipes | 96.3 | 97.24 |
GCA_021605075.1 | Caerostris darwini | 95.6 | 96.66 |
Understanding the Analysis
The workflow, implemented through WDL (Workflow Description Language), requires just two inputs: a list of NCBI accessions and the specific BUSCO lineage for the intended assessment.
Initially, it downloads the lineage file and gets the genome FASTA ftp URL corresponding to the provided accessions.
Subsequently, it downloads the genome from each FASTA URL and initiates compleasm. For a detailed view of the complete workflow, please refer to the GitHub gist provided below.
Hope you liked it! 😃
Subscribe to my newsletter
Read articles from Lucas Taniguti directly inside your inbox. Subscribe to the newsletter, and don't miss out.
Written by
Lucas Taniguti
Lucas Taniguti
I am a bioinformatician and backend developer at Mendelics, a leading genetic diagnostic company in Latin America.