Molecular Docking : I did it
 Biohacker0
Biohacker0Table of contents

finally, I was able to do molecular docking of pantoprazole on the proton pump.
I have so many things I wanna share that I learned from this experience
This article is about what results I got, to know what steps I followed follow this article, it mentions all the steps to be followed to do molecular docking:Blog
Softwares Used: AutoDock Vina, Open Babel, PyMol
1-Open Babel
Open Babel is a versatile chemical file converter that can be used to convert between a wide variety of formats, including sdf, pdb, and pdbqt.In the context of docking pantoprazole to the proton pump, Open Babel can be used to convert the 3D sdf file of pantoprazole to a .pdbqt file.PDBQT files are required by AutoDock Vina, so this is an essential step in the docking process.
2-AutoDock Vina
AutoDock Vina is a molecular docking program that predicts the binding conformation of a ligand molecule to a target protein.It is a popular choice for docking studies due to its speed and accuracy.In the context of docking pantoprazole to the proton pump, AutoDock Vina can be used for the following tasks:Ligand and Receptor Preparation:
AutoDock Vina is instrumental in preparing both the ligand (in this case, pantoprazole) and the receptor (proton pump). It allows for the conversion of these molecules into suitable formats for docking simulations.Grid Setup:
AutoDock Vina assists in defining the grid box, a critical step in molecular docking. This grid box specifies the region within the protein's binding site where docking software explores potential binding poses for the ligand.Docking Simulations:
AutoDock Vina executes the molecular docking simulations, predicting the binding affinity and orientation of the ligand within the receptor's binding site.Analysis:
After docking, AutoDock Vina provides valuable data on binding affinities and potential binding poses, facilitating the identification of favorable interactions.
3-PyMOL
PyMOL is a molecular visualization program that can be used to view and analyze molecular structures.In the context of docking pantoprazole to the proton pump, PyMOL can be used to visualize the ligand, receptor, and final docked output file.This can help understand the binding mode of pantoprazole to the proton pump and identify any potential clashes or steric hindrances.

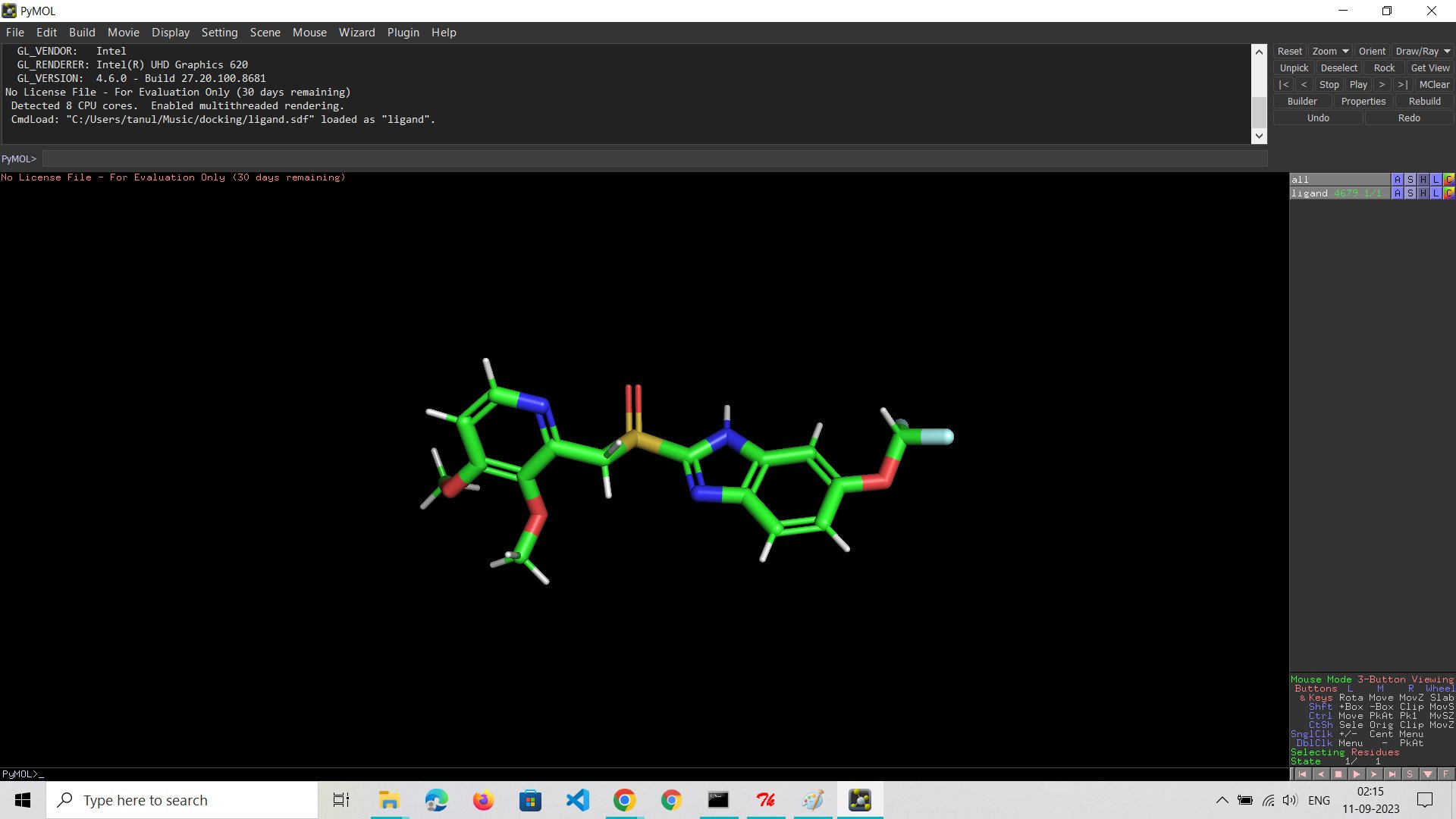
Introduction:
Molecular docking is a powerful tool in the world of drug discovery. It helps us understand how drugs interact with proteins at a molecular level. In this post, I'll share my recent experience with molecular docking, focusing on pantoprazole and proton pumps. We'll discuss the tools used, some limitations, and key findings.
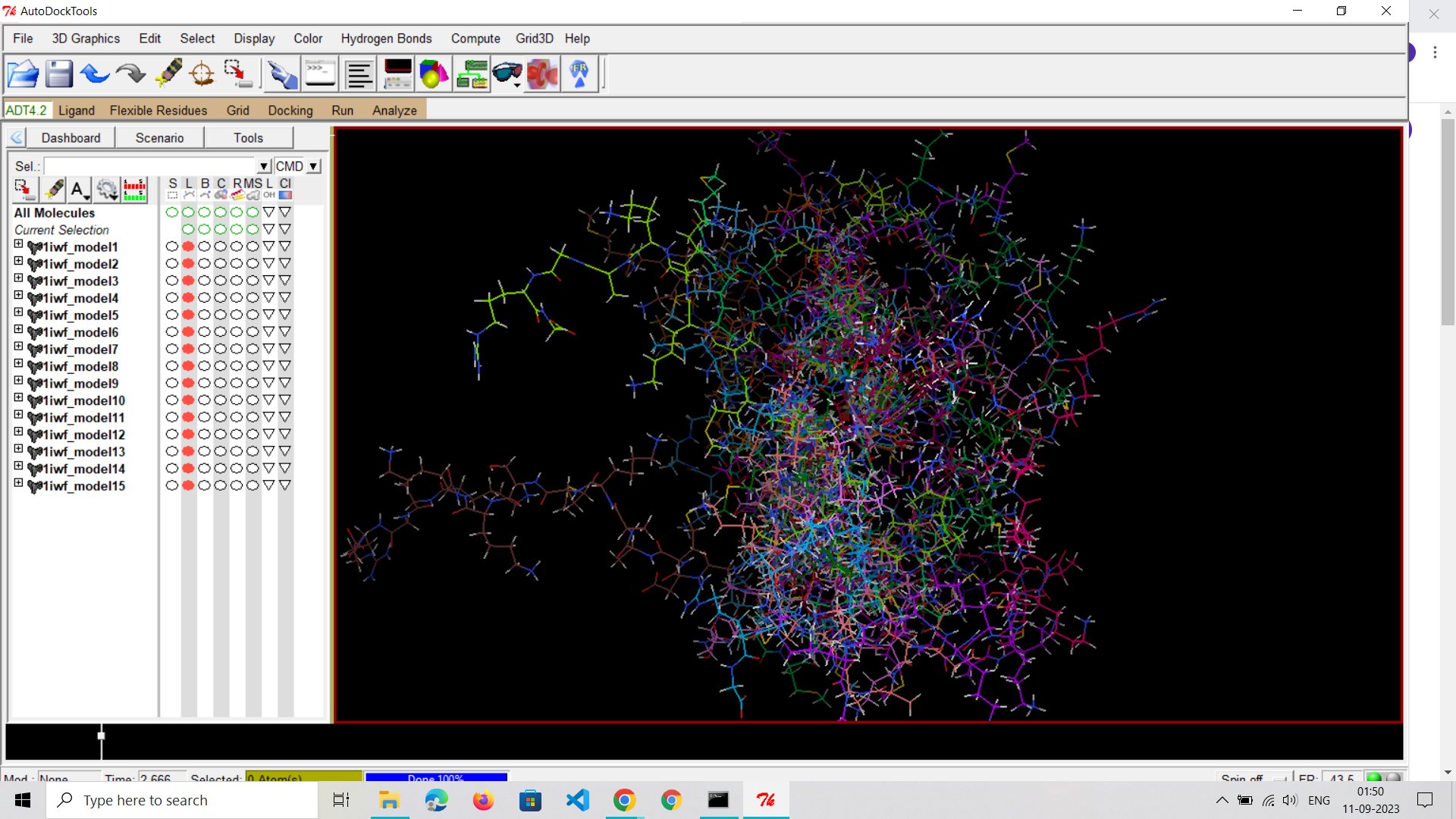
Starting with Autodock Vina: I began my journey with molecular docking using Autodock Vina's user-friendly graphical interface. It's a great place to start for those new to the field.

Model-1 Exploration: I kicked off the study by working with the first of ten models. However, I soon realized that the graphical interface had limitations. To gain more control and precision, I will be shifting to terminal mode and use Python to automate this stuff in future.
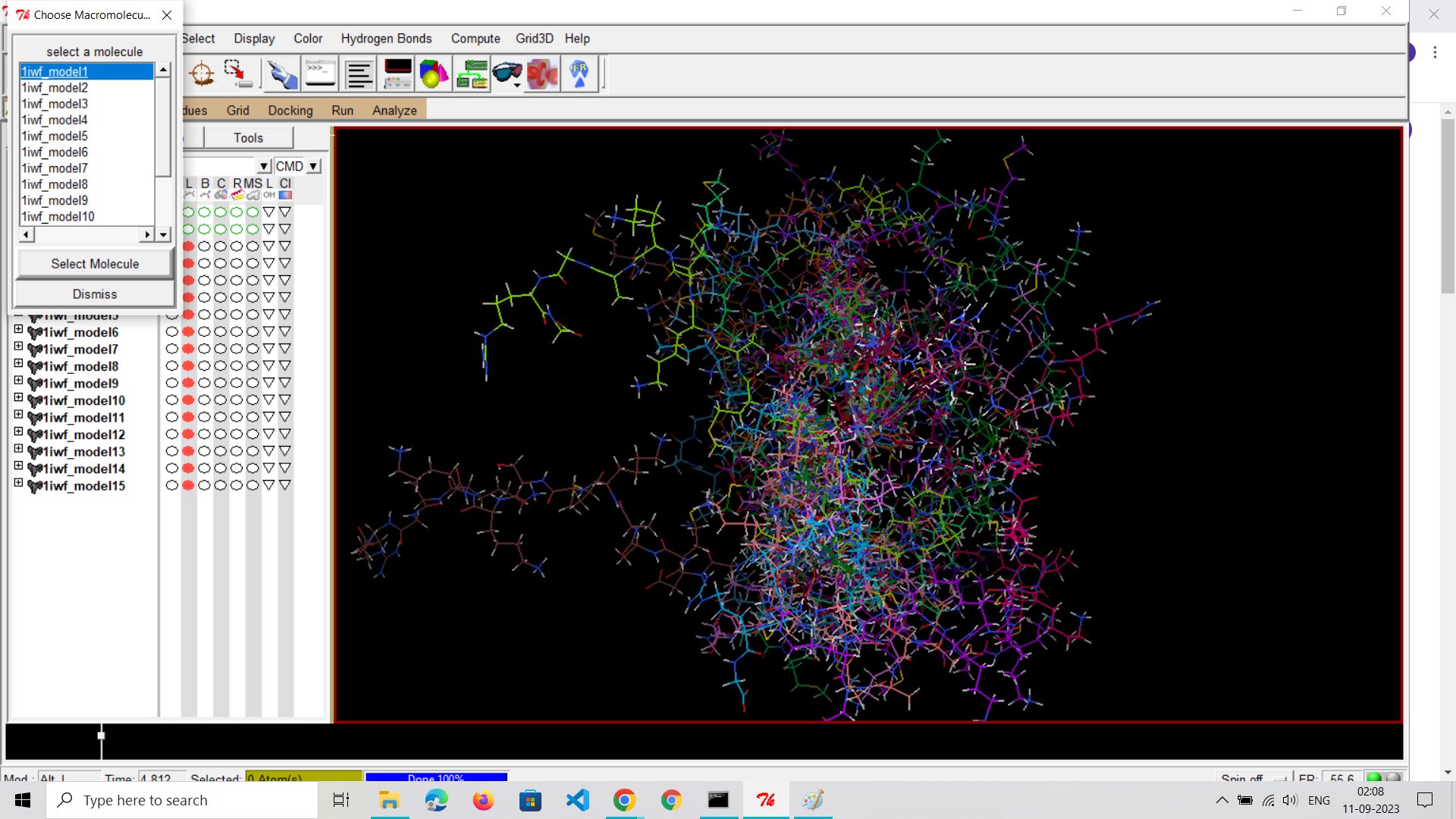
The Importance of the Grid Box: One crucial element in molecular docking is the grid box. This defines the area within the protein's binding site where the software searches for possible drug-binding positions. Getting the position of this box right is critical for the study's accuracy.

Challenges with Coordinates: Finding the exact coordinates for the docking experiment is like trying to hit the bullseye. While there are tools to help, it's a challenging task. None of these tools can guarantee a perfect solution, making molecular docking a complex but fascinating field.

Result:
The Setup: Before delving into the results, it's important to understand the groundwork. The selection of binding sites and the number of trials are determined by the parameters set within the grid box configuration file. This file outlines the specific locations where the docking software attempts to find the optimal binding poses.
Testing with Smaller Parameters: To manage the computational load, I began testing with smaller parameters. This decision allowed for more manageable processing times. However, I plan to extend these experiments to their maximum values in the future, once the feasibility of the current setup is established.
Affinity Scores and Binding Energies: In molecular docking, affinity scores represent the predicted binding energies between pantoprazole and the protein. It's crucial to note that lower (more negative) scores generally indicate stronger binding affinity. The goal is to identify binding sites with the lowest scores, as they are likely to be the most promising for drug-protein interactions. Furthermore, it's essential to consider the distances between the ligand and the protein, favoring smaller values for optimal binding.
Model-1: A Promising Start: In my preliminary tests, I focused on model-1 out of 15 models using a specific set of coordinates and smaller parameters. Surprisingly, this initial run exhibited the best chances of favorable binding interactions. It's worth pondering the vast number of permutations and combinations that can be explored with different models, coordinates, and parameters to obtain the most optimal results.
Output Data:
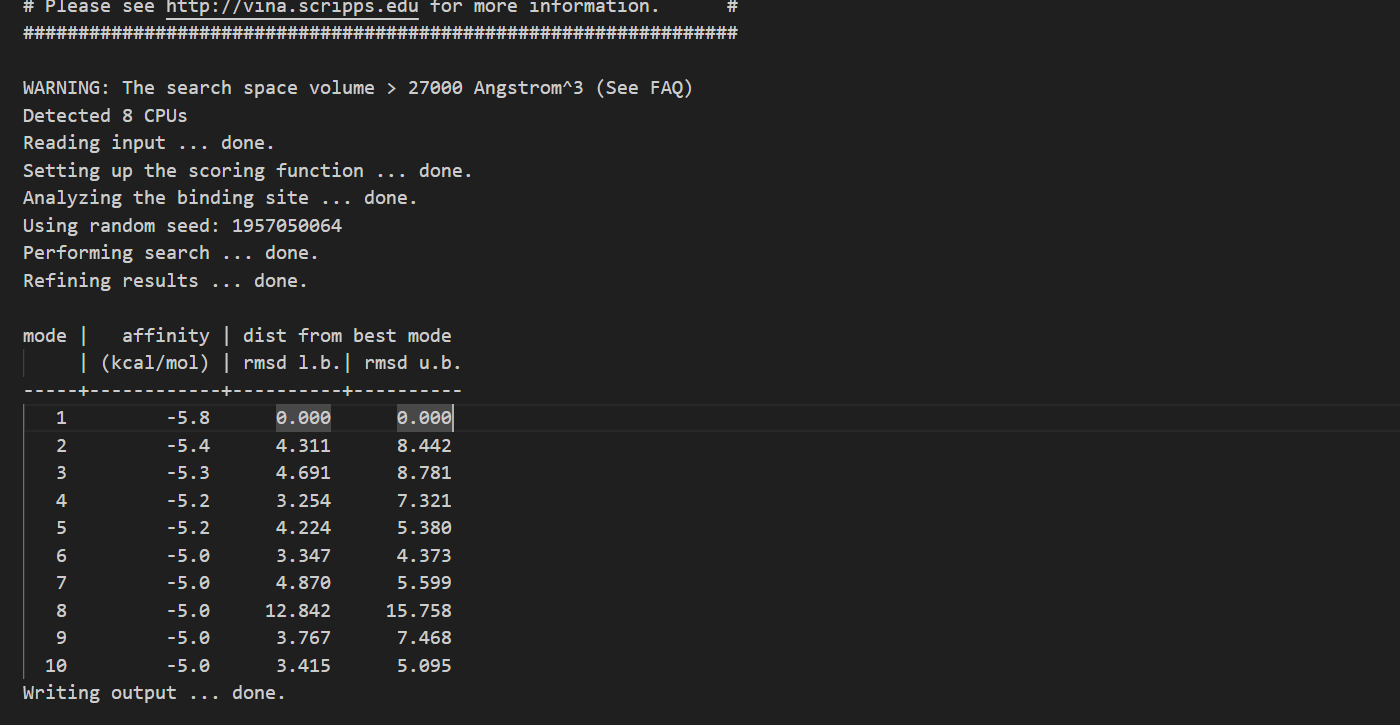
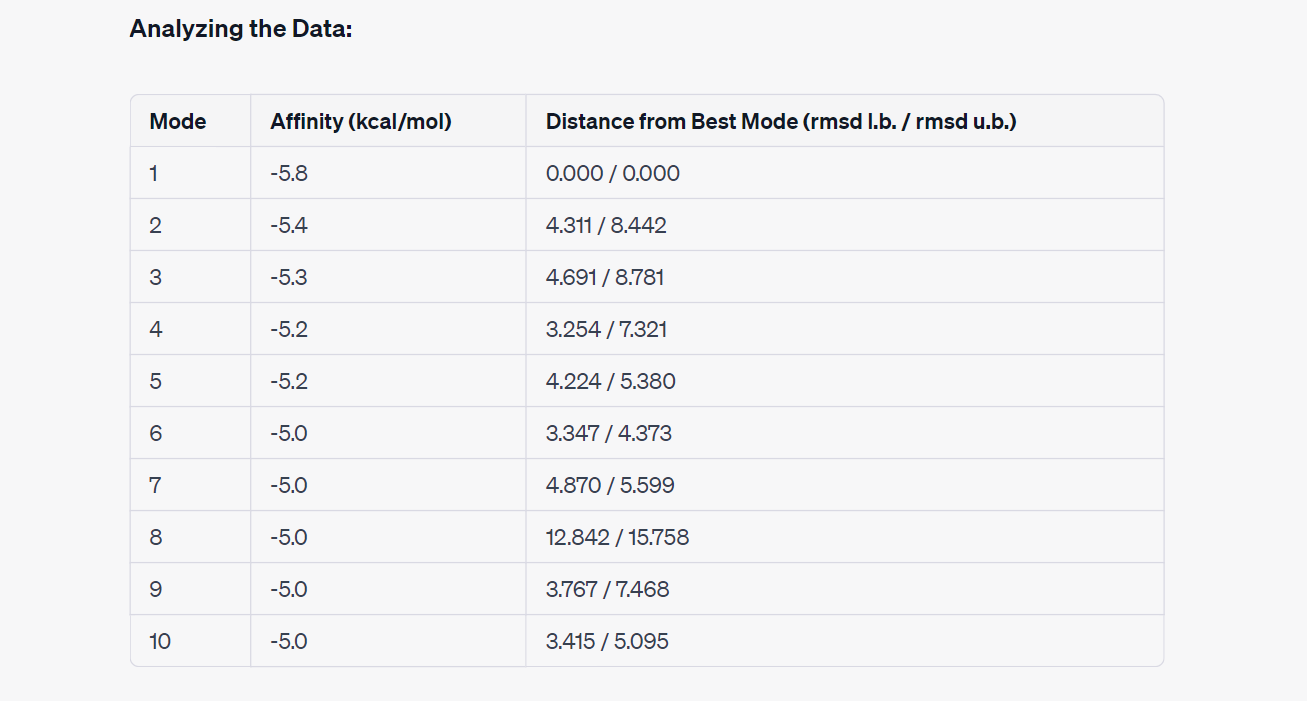
Interpreting the Results:
Mode 1: This mode exhibits the lowest affinity score at -5.8 kcal/mol, making it the most energetically favorable binding pose. It serves as a reference for comparison.
Modes 2 to 7: These modes still demonstrate reasonably strong binding affinities, ranging from -5.4 to -5.0 kcal/mol. However, they are associated with varying distances from the best mode. Modes 2 and 3 have relatively higher distances, suggesting slightly less optimal binding poses.
Modes 8 to 10: In these modes, the affinity scores remain at -5.0 kcal/mol, but the distances from the best mode significantly increase. Mode 8, in particular, has a substantial distance, indicating a less favorable binding configuration.
For Video read this tweet
Remarks/Conclusion:
Since a GUI is not feasible due to the multiple models and parameter permutations, I will write Python scripts to automate the manual tasks I performed in AutoDock. The strategy is to use brute force to try all models with different configurations.
Subscribe to my newsletter
Read articles from Biohacker0 directly inside your inbox. Subscribe to the newsletter, and don't miss out.
Written by
Biohacker0
Biohacker0
Making cool stuff