Getting Started with Biopython Library (Part 1: Obtaining a Sample DNA Sequence)
 Bryan Castillo
Bryan Castillo
When it comes to learning the essentials of computational biology, or what we today refer to as "bioinformatics," Biopython is a very flexible Python library that can help life science enthusiasts and researchers to implement biological data retrieval, manipulation, and analysis. Here's a quick, beginner-friendly guide to get you started on your exciting new path in bioinformatics.
Setting Up the Virtual Environment
To get started, I assumed that you already set up your working environment in any Python compatible integrated development environments (IDEs). If not, I suggest that you can use IDEs such as PyCharm, Spyder, Visual Studio Code (VSCode), Google Collab, or a Jupyter Notebook.
pip install biopython
# to verify if Biopython is installed
pip show biopython
Biopython Import Statement
For this starting tutorial, we need to import the following Biopython classes. Even though Entrez.email can be technically considered as an optional parameter, it is still highly suggested to be included so that the National Center for Biotechnology Information (NCBI) ,which is one of the known service providers of tools and resources in molecular biology, would be able to monitor our usage and can contact us in case of some issues may arise.
from Bio import Seq, SeqRecord # for handling sequences and its record
from Bio import Entrez # for NCBI Database a ccess
Entrez.email = "example@gmail.com"
Other Biopython classes that can be used based on the documentation includes:
from Bio import SeqIO, AlignIO # working with sequence alignment
from Bio import PopGen # for population genetics like allele statistics
from Bio import PDB # for protein database and protein related analysis
Obtaining a Sample DNA Sequence
Method 1 : Simple Text (String)
This part doesn't use the Biopython library yet. Creating a simple nucleotide sequence from a randomly generated string representing the nucleotide bases. This uses the built-in random module, and using the join method to create some strings from the given list. The length of your desired sequence, you can change the value in the range method. For this example, I chose a sequence length of 50.
import random
Nucleotide = ["G","T","A","C"]
DNA_string = "".join([random.choice(Nucleotide) for nuc in range(50)])
DNA_string
Example Output: 'CTGGGTTGTAGAACCAATGAACAGAGCATGGCTGATAATGTATCCGAGCA'
Method 2: Parsing a URL
In this part, we are going to deal with an actual DNA sequence that can be retrieved from the NCBI Database. There are three functions created to provide a clean and step by step process of retrieving a desired DNA sequence. Assume that you just get the URL of the NCBI web page and want to retrieve the sequence on the NCBI web page.
from urllib.parse import urlparse
def extract_accession_number(url):
segments = urlparse(url).path.split("/")
accession_number = segments[-1]
return accession_number
def getnuc_from_ncbi(url):
accession_number = extract_accession_number(url)
if not accession_number:
print("Invalid URL. Please provide a valid NCBI nucleotide sequence URL.")
return
handle = Entrez.efetch(db="nucleotide", id=accession_number, rettype="gb", retmode="text")
record = SeqIO.read(handle, "genbank")
handle.close()
return record
def save_fasta(record):
with open("output_seq.fasta", "w") as f:
fasta_format = f">{record.id} {record.description}\n{record.seq}"
f.write(fasta_format)
I think you can easily comprehend how the first function works if you have some basic Python knowledge. If not, it simply uses the urlparse function from Python's built-in urllib.parse package to get the URL. The next step is to divide the URL into segments using the forward slash character as the indicator of splitting. The next action is to slice the string in which the last string typically contains the accession number, may be captured by stating the [-1] position.
Accession number is esentially a unique identifier in a biological database by which it is open to the public or shared through scientific research papers to allow access on fellow researchers. This also helps in collaborations and result validations of the provided data in the research involved within the scientific community.
After that, the second function (getnuc_from_ncbi) is for the data retrieval part. The handle object stores the fetched data using the efetch method from the Entrez module. As you can see on the db parameter, it was defined with "Nucleotide" which pertains to what specific database of the NCBI. Since we are focused on obtaining the nucleotide or DNA sequence, we could recover it from the Nucleotide database. Meanwhile, the rettype parameter specifies what is the data type to be fetched while retmode which stands for retrieval mode is set to "text" to allow a human readable format. In this case, it is the Genbank format, which is a format of DNA sequence data type that contains not only the sequence itself but some other relevant informations.
The next line is for SeqIO.read is the method to read a single sequence in a format distinguishable by the Biopython because the handle object which was freshly fetched from the NCBI database is not yet ready for further processing associated in the Biopython library. Finally, the handle.close() line of code is a disciplinary way to close any database operation such as the fetching of the data. This is mainly for resource clean up and a good practice to add at the end. Moreover, the concept is similar on how you perform some MySQL operations in Python.
The last function which is save_fasta with the parameter record generally allows us to save the recovered Genbank to data to fasta format. In bioinformatics, fasta file format is the most simple file to store nucleotide or protein sequences. To save it, this code utilizes the with open statement to read and write the record object allowing us to structure the content of the fasta file. As those lines of code were clearly explained, let us now demonstrate the usage of those functions:
# Example Usage
ncbi_url = "https://www.ncbi.nlm.nih.gov/nuccore/L08050.1"
record = getnuc_from_ncbi(ncbi_url)
save_fasta(record)
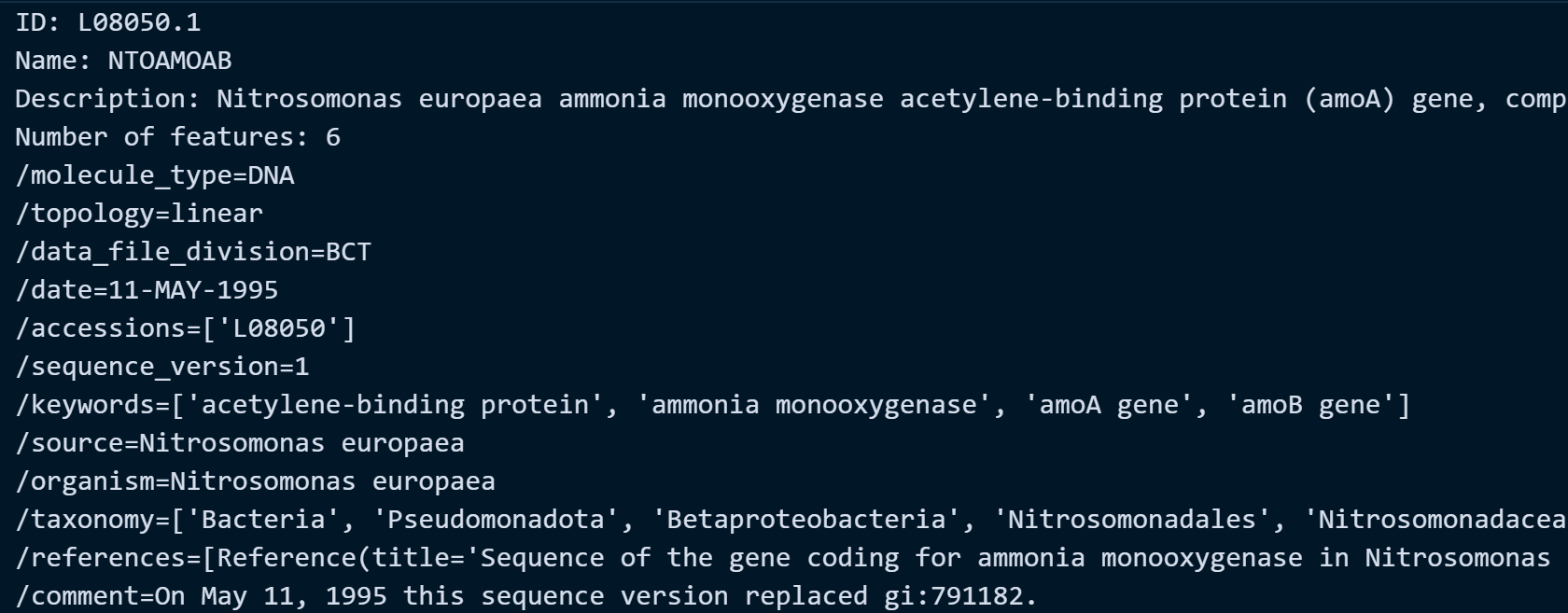
print(record)
Let's try the URL provided on ncbi_url object. In this example, we are fetching the amoA gene of Nitrosomonas europaea with accession ID: L08050.1 (a type of ammonia-oxidizing bacteria that has a functional gene amoA responsible for converting ammonia into nitrite in the nitrogen cycle of aquatic environment). These bacterial species were commonly observed and studied on biological wastewater treatment systems.
Example Output:
Method 3: Reading a Text File as CSV File Format containing the Accession ID and the Name of the Species
This scenario is wherein that you already have a list of accession IDs of the nucleotide sequence with the name of the species associated with it. For example, you have this .txt file in this comma-separated format:
AF313802.1,Methanosaeta concilii strain DSM 3671
AF414043.1,Methanosarcina mazei strain DSM 2053
AY970349.1,Methanosaeta harundinacea strain 6Ac
AY970348.1,Methanosaeta harundinacea strain 8Ac
AY672820.1,Methanomethylovorans thermophila
AF414045.1,Methanocorpusculum parvum strain DSM 3823
KC876049.1,Methanomethylovorans uponensis strain EK1
GU385700.1,Methanobrevibacter smithii strain 4R_4_F02
KF836868.1,Uncultured Methanoculleus sp. clone HB-107
KF836855.1,Uncultured Methanoculleus sp. clone XJ6-100
As you can observe from this snippet, we have the accession numbers then followed by a comma and then the name of the species. For this example, let us save the list as methanogens.txt. For the biological context, these group of archaeal species were the main methane producing microorganisms in the anaerobic digestion process, a popular biological process for organic waste degradation in the environment.
We can fetch each of those species on the list again with the efetch method and then work on organizing the format to achieve an easy-to-read format like the fasta file. Here is the code on how it works:
import csv
from Bio import SeqIO
from Bio import Entrez
fasta_file = {}
ct = 0
with open('methanogens.txt') as csv_file:
csv_reader = csv.reader(csv_file, delimiter=',')
for row in csv_reader:
print(row)
with Entrez.efetch(db="nucleotide", rettype="gb", retmode = "text", id = row[0]) as handle:
for seq_record in SeqIO.parse(handle, "gb"):
accession_number = seq_record.annotations['accessions'][0]
header = f"> {accession_number} {row[1]}"
fasta_file[header] = str(seq_record.seq)
ct += 1
# This print statement notifies you on the content of the file and how many sequences can be retrieved
print("Number of sequences in file:", ct)
# Saving the output in fasta format
with open("sequences_general.fasta","w+") as f:
for header, sequence in fasta_file.items():
f.write(f"{header}\n{sequence}\n")
Upon reading the .txt file, it reads the file in comma-separated-value format followed by iterating the rows to perform the fetching. It has the same process as mentioned on Method 2 but this time, we are trying to fetch multiple sequences using the provided list as the reference to look upon. As you can see in the code, it is a nested for loop to continue iterating each element to structure the content of the fasta format and then the outer for loop is for the iteration of reading each row from the list provided in the .txt file.
The fasta_file dictionary is created to store the retrieved sequences in FASTA format. Each entry in the dictionary has a header composed of the accession number and additional information from the CSV file. Meanwhile, the ct variable keeps track of the total number of sequences retrieved. Both fasta_file and ct are initialized outside the nested loops to allow the storage of data during the iterative process. The final print statement informs the user about the content of the file and how many sequences were successfully retrieved.
Method 4: Reading a Fasta File Containing Multiple Sequences and Saving as a CSV File Format with relevant Informations
This approach works well in the case your original data source is a fasta file with several sequences. This method of obtaining DNA sequence would use pandas library which is a popular data handling tool in Python. Together with Biopython, we can organize a fetched data into a readable dataframe of containing the accession ID, name of the species, sequence, and the sequence length. Here is the code snippet for the first part (fetching of the sequence data and filling up the desired lists of target information):
from Bio import SeqIO
import pandas as pd
with open("sequences_general.fasta",'r') as fasta_f:
id, description, sequence, lengths = [], [], [], []
for seq_record in SeqIO.parse(fasta_f,'fasta'):
id.append(str(seq_record.id))
description.append(str(seq_record.description))
sequence.append(str(seq_record.seq))
lengths.append(len(seq_record.seq))
The code begins with reading a fasta file. For this tutorial, we can use the output from method 3 (sequences_general.fasta) as our example input in this approach. To store the information retrieved during iteration, four empty lists (id, description, sequence, and lengths) are created. The Biopython SeqIO.parse method is utilized to efficiently read the FASTA file and extract specific information about each sequence. The append method is then used to populate the lists with the relevant details in the order of ID, description, sequence, and length.
df = pd.DataFrame({'Accession ID': id, 'Name': description,
'Sequence': sequence,'Length': lengths })
df['Name'] = df['Name'].str.join('').str[10:]
df.head(10)
Subsequently, the Pandas library is leveraged to create a DataFrame. Using the pd.DataFrame method and a dictionary, each key represents a column in the DataFrame, and the corresponding values are the lists formed during the iteration with SeqIO.parse. Finally, some modifications were made to the 'Name' column by removing leading whitespaces found towards index 10. Organizing the dataframe could result to an output shown below:
| Accession ID | Name | Sequence | Length |
| AF313802 | Methanosaeta concilii strain DSM 3671 | GGGAAGCTACATGTCCGGCGGTGTCGGTTTCACCCAGTACGCTACT... | 472 |
| AF414043 | Methanosarcina mazei strain DSM 2053 | TACACAGACGACATCCTCGACAACAACACCTACTATGACGTTGACT... | 435 |
| AY970349 | Methanosaeta harundinacea strain 6Ac | ACGTTCATGGCGTAGTTCGGATAGTTCGCGCCTCTCAGCTCGAGGG... | 507 |
| AY970348 | Methanosaeta harundinacea strain 8Ac | ACGTTCATGGCGTAGTTCGGATAGTTCGCGCCTCTCAGCTCGAGGG... | 507 |
| AY672820 | Methanomethylovorans thermophila | TAGGTCCACGCAGCTCAAGAGGCAGACCTTCGTCGGACTGGTAGGA... | 739 |
| AF414045 | Methanocorpusculum parvum strain DSM 3823 | TACACGGATAACATCCTTGATGACTTCACCTACTACGGAATGGACT... | 438 |
| KC876049 | Methanomethylovorans uponensis strain EK1 | AGCATGTGTGCCGGTGAAGCAGCAGTCGCTGACCTTTCATATGCAG... | 706 |
| GU385700 | Methanobrevibacter smithii strain 4R_4_F02 | ATCTTCAAATTTCTGATGGAGAATACGTTAGCTGCACCACATTGAT... | 423 |
| KF836868 | Uncultured Methanoculleus sp. clone HB-107 | GGCATACACCGACAACATCCTGGATGAGTTCACATACTACGGTATG... | 436 |
| KF836855 | Uncultured Methanoculleus sp. clone XJ6-100 | GGCCTACACCGACAACATCCTCGATGAGTTCACCTACTACGGTATG... | 436 |
Conclusion
These methods represent just a glimpse into the versatile ways Biopython empowers researchers to organize and retrieve DNA sequences for further analysis. The Biopython library, with its rich set of tools, offers a plethora of possibilities for biological exploration and computational biology. In this lesson, we've scratched the surface, delving into various techniques to harness the power of Biopython.
For those eager to explore more of the functionalities and dive deeper into the world of bioinformatics with Biopython, stay tuned! Future blog posts will continue to showcase the diverse capabilities of this library. Whether you're a seasoned bioinformatician or just starting your journey, Biopython is a powerful ally, providing the tools needed to unravel the mysteries encoded in DNA sequences.
Continue exploring, stay curious, and let the fascinating world of computational biology unfold before you.
Subscribe to my newsletter
Read articles from Bryan Castillo directly inside your inbox. Subscribe to the newsletter, and don't miss out.
Written by
Bryan Castillo
Bryan Castillo
A researcher with a background in Biological Engineering, Chemistry, and Environmental Engineering. Integrating my skills with programming and new tech stacks.