Lab Notes: Building a GenePT Cell Typer - Part 1
 RJ Honicky
RJ Honicky
Introduction
We recently built a cell-typer model based on a subset of the CellXGene V2 dataset. This model is a stepping stone towards a “universal” cell-typer model that will allow us to annotate cell types (and other properties) based on single cell RNA transcription data.
I’ve noticed that people prefer shorter, multi-part articles over longer ones, so I’ve broken these Lab Notes up into three parts.
Part 1: CellXGene V2 EDA and test set filtering ← you are here
Part 2: Training and test set construction
Part 3: Model design and training
If you’re interested in more detail, you can find the notebook I used for the EDA here: https://github.com/honicky/GenePT-tools/blob/main/notebooks/cellxgene_v2_training_data_eda.ipynb
I’ll publish Part 2 and Part 3 as quickly as I can write them up!
Data curation
Despite attempts at generalization, existing cell-typing mechanisms such as Census require relatively complex setup and also fine-tuning to a particular data set or cell class. Recent Transformer-based single-cell RNA “foundation” models still require technical complexity and fine tuning to work well for specific problems. On the other hand, we believe that they are a big step on the path towards a simple “universal” cell-typing workflow in which a user can input their scRNA dataset and the model outputs a labeled set without fine tuning or setup.
GenePT and scGPT are two models that use complementary approaches and combining them together has already shown promising academic results for gene perturbation analysis. I am working with Miraomics and Pythia Biosciences to explore a similar approach for cell-type annotation. To start with, we are building a small training and test set in order to understand the training process for a “stacked” scGPT and GenePT model. Understanding how to build a good training and test set is usually one of the hardest parts of building a good model, so I’m documenting the process we followed in detail so that anyone can reproduce what we’ve done and improve on it.
anndata-metadata - A little tool for reviewing metadata
Pythia and Miraomics have collected the large open-source CellXGene V2 dataset published by the CZ institute into an S3 bucket which makes the curation process way easier for me. You can download the dataset yourself at https://cellxgene.cziscience.com/datasets.
On the other hand, I still want an easy way to understand the statistics of the whole dataset, file-by-file, so I created a little utility to crawl over each file, extract some of the key metadata and then output it to a parquet file. This enabled a lot of the analysis that follows, as well as ad-hoc queries using duckdb / pandas / whatever. You can find the tool at
https://github.com/honicky/anndata-metadata
I used this to crawl the S3 bucket with the AnnData files from CellXGene V2 and extract information about the cell-type distribution. Here are the top 20 most prevalent cell type labels in the dataset:

And the full set

Just use (almost) everything…
We originally assumed we would filter out cells and files that had abnormal read-count or gene-count distributions. Using our metadata tools, we learned that the different files have different names for the read-count (meaning the total number of genes of any type detected per cell) and the gene-count (meaning the total number of distinct genes detected per cell). We visually inspected these, and came up with the following list:
# 1. List of read counts (including UMIs)
read_count_cols = [
'Number of UMIs',
# 'nCount_ADT',
# 'nCount_Exon',
# 'nCount_HTO',
'nCount_RNA',
'nCount_SCT',
'nCount_originalexp',
'nUMI',
'nUMIs',
'n_counts',
'n_counts_UMIs',
'total_UMIs',
'total_counts',
'total_transcript_counts',
'n_umis',
'sct_n_umis',
'gene.counts',
'n_count_rna',
'ncount_rna',
]
# 2. List of gene counts (non-zero reads)
gene_count_cols = [
'Genes detected',
'Genes.Detected',
'Genes.Detected.CPM',
'detected',
'genes',
'nFeature_RNA',
'nFeature_SCT',
'nGene',
'n_feature_rna',
'n_features',
'n_genes',
'n_genes_by_counts',
'n_genes_detected',
'nfeature_rna',
'ngenes',
# 'sct_n_genes',
'total_features',
'total_features_by_counts',
'total_genes'
]
Using this list, we can review the distribution the number of reads and genes per cell for each file as a heatmap. Here are truncated versions (check out the notebook for the full images):
Read count histograms
Gene Count histograms
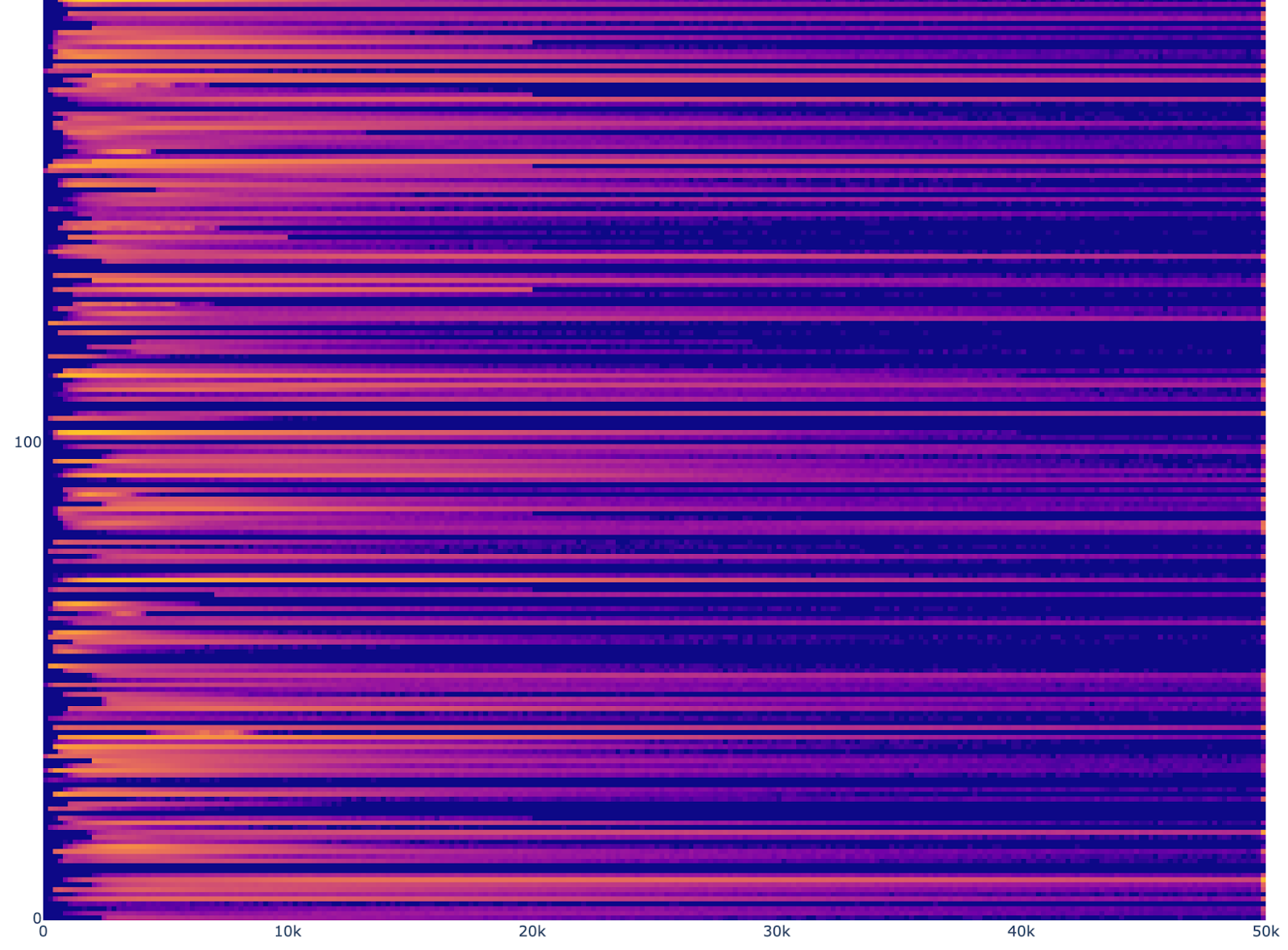
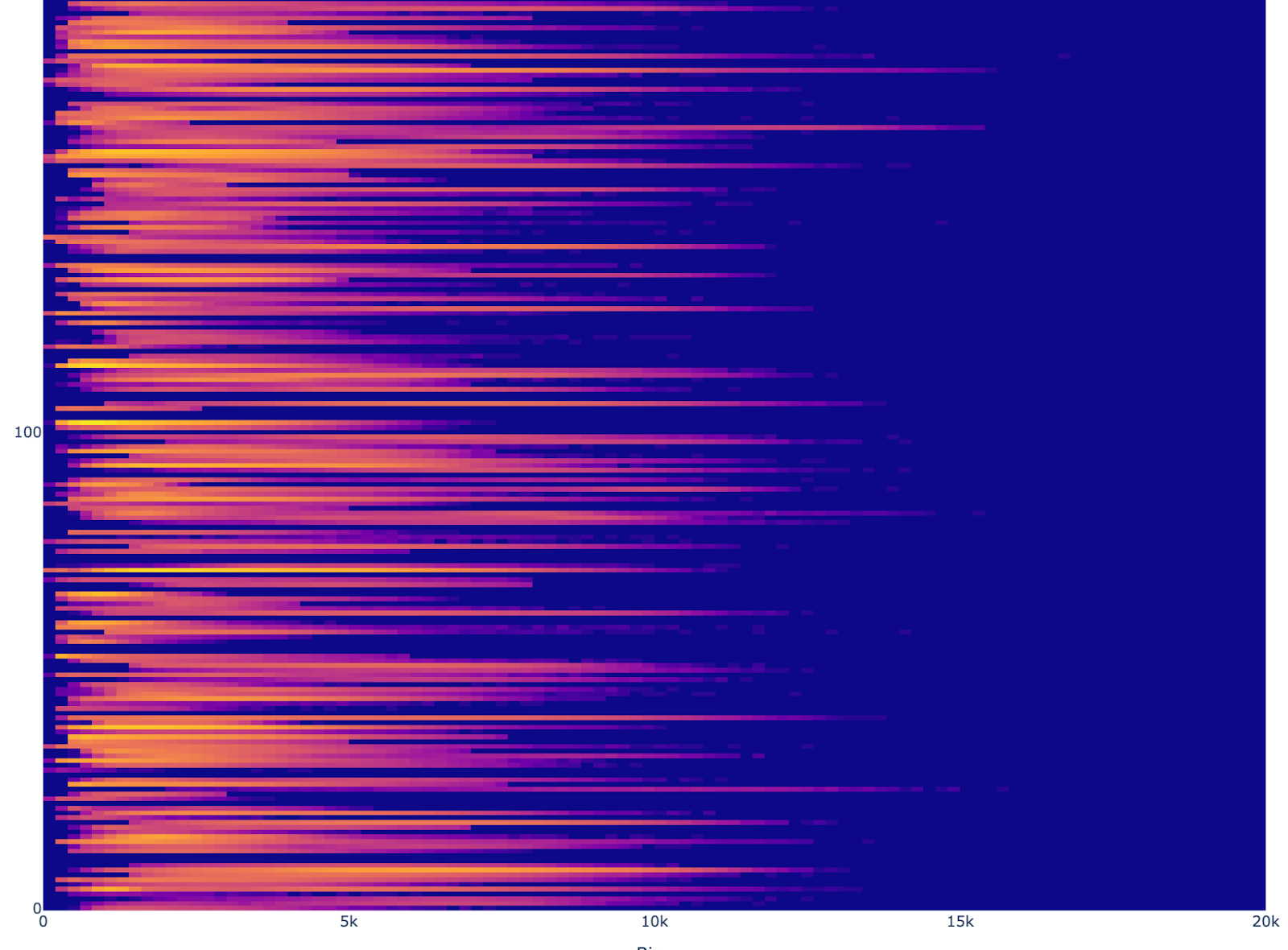
The read counts have a great deal of variability. One source of this variability in distribution is assay. We will include other assays in future models, but in order to simplify the analysis, we chose to train only on 10X for this initial model.
We also decided that it would be difficult to justify any particular threshold, so we decided to rely on the variability of the data to span the distribution of possible configurations and average out the abnormal readings. This introduces the risk that outliers and low-read-count data will impact performance. We have chosen to start without extensive filtering so that we can build a model, with the goal of using the model itself to help with filtering via perplexity or other confidence-related metrics. The upshot is that we filter by assay and cell types with adequate representation, but otherwise use all files and cells.
Test set - after the scGPT training date (2023-05-08)
The exact training set for scGPT is a bit unclear, so in order to ensure that we do not validate using data from the training set, we only use files that were curated after the scGPT training date for the test set (2023-05-08).. This includes 393 of the 961 total files available to be included in the test set, and that 152 of the total 833 cell types are not represented in the test set.
This also means that we do not capture advances in sequencing quality that happened after the cutoff date. This makes the training and test sets good for capturing the robustness of the algorithm to changes in sequencing technology and process over time, but probably not as up-to-date as it could be. After testing with GenePT and scGPT together, we will re-training using a more modern dataset.
Next up: overcoming IO constraints to build a test set
The CellXGene v2 data set we are using is 3.14TB, and only a small subset of the data from these files - stored in S3 - are actually relevant for training our models. This makes the IO problem an interesting and tricky one, so I’ll write up the tricks I used to keep the IO managable.
As always, your questions and feedback, especially negative, is greatly appreciated. Find me on
LinkedIn: rj-honicky
BlueSky: honicky.bsky.social
X: honicky
Subscribe to my newsletter
Read articles from RJ Honicky directly inside your inbox. Subscribe to the newsletter, and don't miss out.
Written by